Advanced probeset selection
In this tutorial we look into customizing the gene set selection for more specific use cases and explore the parameters of the selection procedure.
[3]:
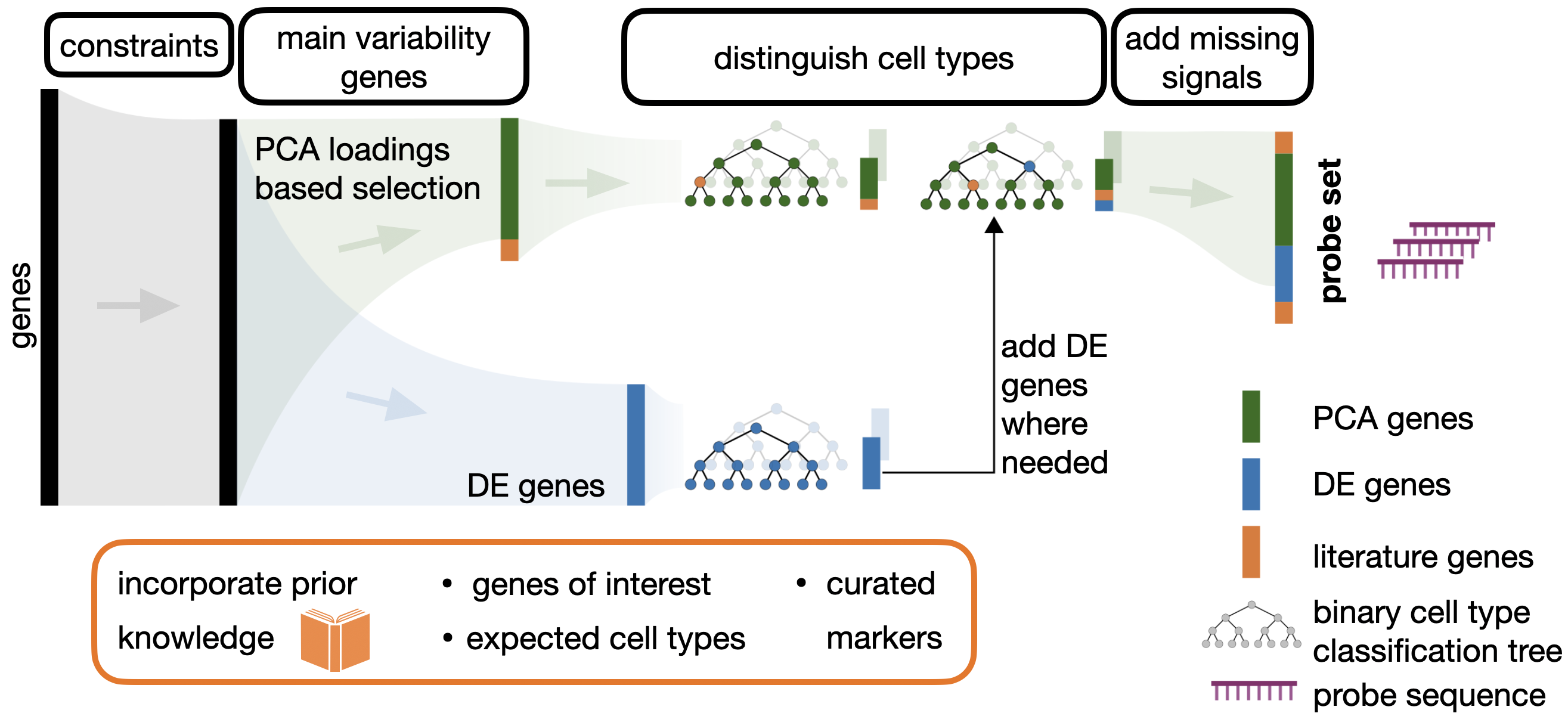
The Spapros probeset selection pipeline simultaneously optimizes for capturing transcriptional variation and cell type identification while taking into account prior knowledge, technical constraints and probe design. The main steps, as visualized in the figure above, are:
Constrains
In the first step, genes are filtered based on technology specific probe design constraints. This part is captured in the end-to-end probeset selection tutorial. (Besides probe design constraints also expression level constraints can be applied as shown in the Selection with experession constraints example below. Those affect multiple steps of the pipeline though)
Variable genes
The next step is a PCA based selection of genes that capture general variation. The subsequent steps of the pipeline are encouraged to select from that set of genes. If you want the selection to be focused only on marker genes and cell type classification, the PCA-based selection step can be omitted, see the Cell type classification only example for this case.
Distinguish celltypes
For each cell type, a binary classification tree is trained on the previously selected gene set. Here the pipeline builds for each cell type a combinatorial rule how to distinguish it from other cell types.
DE gene addition
Similarly to the last step, trees are also trained on DE genes and compared to the trees trained on the PCA based selection. If a DE tree performs better with respect to cell type classification, then DE genes are added to the already selected gene set.
Prior knowledge
There are two possibilities to incorporate prior knowledge. For both, we present use cases in this tutorial:
Select a few additional genes: pre-selected genes are augmented by combinatorially selecting additional genes around them
Selection with curated marker list: add markers from a given list in case the signals from that list are not captured with the already selected genes
Probe design
Design the final technology specific probes for the selected genes. As the Constraints step, this part is also only captured in the end-to-end probeset selection tutorial.
Content
-
Selection for high numbers of genes (> 150)
Your use case is not captured?
What’s next
0. Import packages
[4]:
import numpy as np
import scanpy as sc
import spapros as sp
[5]:
sc.settings.verbosity = 0
sc.logging.print_header()
print(f"spapros=={sp.__version__}")
scanpy==1.9.1 anndata==0.8.0 umap==0.5.3 numpy==1.21.6 scipy==1.7.3 pandas==1.3.5 scikit-learn==1.0.2 statsmodels==0.13.2 python-igraph==0.9.11 pynndescent==0.5.7
spapros==0.1.0
1. Load and preprocess data
For Spapros selections the count data should be log-normalised. Notably genes should not be scaled to mean=0 and std=1. We pre-select a given number of highly variable genes, here 1000. In real world applications we typically go for 8000.
The example data set that we use here has a raw version and a processed version with scaled counts. To get log normalised counts we use the raw version. Cell and gene filters, cell type annotations, and the umap embedding we get from the processed version.
[6]:
adata = sc.datasets.pbmc3k()
adata_tmp = sc.datasets.pbmc3k_processed()
# Get infos from the processed dataset
adata = adata[adata_tmp.obs_names, adata_tmp.var_names]
adata.obs['celltype'] = adata_tmp.obs['louvain']
adata.obsm['X_umap'] = adata_tmp.obsm['X_umap']
del adata_tmp
# Preprocess counts and get highly variable genes
sc.pp.normalize_total(adata)
sc.pp.log1p(adata)
sc.pp.highly_variable_genes(adata, flavor="cell_ranger", n_top_genes=1000)
adata
[6]:
AnnData object with n_obs × n_vars = 2638 × 1838
obs: 'celltype'
var: 'gene_ids', 'highly_variable', 'means', 'dispersions', 'dispersions_norm'
uns: 'log1p', 'hvg'
obsm: 'X_umap'
2. Use cases
We will go through some specific use cases for gene selection.
i. Select a few additional genes
In some situations you already selected multiple genes that you want to measure in a targeted spatial transcriptomics experiment but you still have capacity for further genes. For this scenario the preselected_genes parameter was introduced.
Let’s distinguish two scenarios
We preselected 40 genes and want to add 10 (adds up to 50 in total)
We preselected 40 genes and want to add 100 (adds up to 140 in total)
For the 2. scenario simply adding the first 40 genes in preselected_genes and going with default parameters is the right way to go. For the 1. scenario (which we look at in the code below) the parameter n_pca_genes should be reduced. The reason is that Spapros will build trees based on the pre selected genes and the genes from the PCA selection. If there are a lot of PCA genes in the pool, then the trees might ignore most of the preselected genes. This is not extremly problematic, but
it’s preferable to build up our combinatorial rules on the pre selected genes. We’ll reduce n_pca_genes from 100 (default) to 20 for the given case. Note however that the preselected_genes can include genes that overlap with the pca genes, therefore we more specifically set n_pca_genes to the number that provides us 20 more pca genes on top of the preselected list.
[7]:
# list of pre selected genes
preselected_genes = [
'PF4', 'HLA-DPB1', 'FCGR3A', 'GZMB', 'CCL5', 'S100A8', 'IL32', 'HLA-DQA1', 'NKG7', 'AIF1', 'CD79A', 'LTB', 'TYROBP',
'HLA-DMA', 'GZMK', 'HLA-DRB1', 'FCN1', 'S100A11', 'GNLY', 'GZMH', 'CST3', 'RBM39', 'HLA-DPA1', 'LST1', 'LGALS2',
'FCER1A', 'MS4A1', 'TRAF3IP3', 'PRF1', 'LGALS1', 'PRDX1', 'ATP5O', 'C1orf162', 'CTSS', 'SELL', 'CTSW', 'GIMAP4',
'LINC00926', 'MS4A6A', 'GSTP1'
]
# Get `n_pca_genes` to obtain the top 20 pca genes that are not already in preselected_genes
tmp = sp.se.select_pca_genes(adata, n=adata.n_vars, inplace=False)["selection_ranking"]
n_pca_genes = tmp.loc[~tmp.index.isin(preselected_genes)].sort_values().iloc[:20].max().astype(int)
# create an instance of the ProbesetSelector class
selector = sp.se.ProbesetSelector(
adata,
n=50,
n_pca_genes=n_pca_genes,
preselected_genes=preselected_genes,
celltype_key="celltype",
verbosity=1,
save_dir=None,
)
# select probe set
selector.select_probeset()
Note: The following celltypes' test set sizes for forest training are below min_test_n (=20):
Dendritic cells : 9
Megakaryocytes : 3
The genes selected for those cell types potentially don't generalize well. Find the genes for each of those cell types in self.genes_of_primary_trees after running self.select_probeset().
Now we can investigate the selected gene set. The annotations in the table describe where genes originate from:
the first 40 genes are annotated with
pre_selected=Truethe added genes from 41 till 50 are annotated with
selection=Trueandpre_selected=False
[8]:
display(selector.probeset.head(4))
display(selector.probeset.iloc[38:42])
display(selector.probeset.iloc[48:52])
| gene_nr | selection | rank | marker_rank | tree_rank | importance_score | pca_score | pre_selected | prior_selected | pca_selected | celltypes_DE_1vsall | celltypes_DE_specific | celltypes_DE | celltypes_marker | list_only_ct_marker | required_marker | required_list_marker | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PF4 | 1 | True | 1.0 | 1.0 | 1.0 | 1.000000 | 0.810399 | True | False | False | Megakaryocytes | Megakaryocytes | Megakaryocytes | False | True | False | |
| CD79A | 2 | True | 1.0 | 1.0 | 1.0 | 0.895466 | 1.226536 | True | False | False | B cells | B cells | B cells | False | True | False | |
| CCL5 | 3 | True | 1.0 | 1.0 | 1.0 | 0.545002 | 2.553322 | True | False | True | CD8 T cells | CD8 T cells | CD8 T cells | False | True | False | |
| GZMB | 4 | True | 1.0 | 1.0 | 1.0 | 0.530616 | 1.252898 | True | False | False | NK cells | NK cells | NK cells | False | True | False |
| gene_nr | selection | rank | marker_rank | tree_rank | importance_score | pca_score | pre_selected | prior_selected | pca_selected | celltypes_DE_1vsall | celltypes_DE_specific | celltypes_DE | celltypes_marker | list_only_ct_marker | required_marker | required_list_marker | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MS4A6A | 39 | True | 1.0 | NaN | 3.0 | 0.003518 | 0.481730 | True | False | False | False | False | False | ||||
| GIMAP4 | 40 | True | 1.0 | NaN | 3.0 | 0.001383 | 2.422619 | True | False | True | False | False | False | ||||
| GPX1 | 41 | True | 2.0 | 2.0 | 1.0 | 0.002822 | 2.025547 | False | False | True | Megakaryocytes | Megakaryocytes | Megakaryocytes | False | True | False | |
| FCER1G | 42 | True | 2.0 | NaN | 1.0 | 0.010767 | 1.324965 | False | False | True | FCGR3A+ Monocytes | FCGR3A+ Monocytes | FCGR3A+ Monocytes | False | False | False |
| gene_nr | selection | rank | marker_rank | tree_rank | importance_score | pca_score | pre_selected | prior_selected | pca_selected | celltypes_DE_1vsall | celltypes_DE_specific | celltypes_DE | celltypes_marker | list_only_ct_marker | required_marker | required_list_marker | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD79B | 49 | True | 3.0 | NaN | 3.0 | 0.022714 | 1.304566 | False | False | True | B cells | B cells | B cells | False | False | False | |
| ARL6IP5 | 50 | True | 4.0 | NaN | 4.0 | 0.016238 | 2.319210 | False | False | True | False | False | False | ||||
| STK17A | 51 | False | 5.0 | NaN | 5.0 | 0.006099 | 1.412628 | False | False | True | False | False | False | ||||
| GIMAP7 | 52 | False | 5.0 | NaN | 5.0 | 0.001398 | 2.961351 | False | False | True | CD4 T cells | CD4 T cells | CD4 T cells | False | False | False |
Note that besides the preselected_genes parameter there is also the prior_genes parameter (for “prioritized genes”). This one acts similar to preselected_genes, however genes in prior_genes are not necessarily selected, they’re only added to the pool of PCA preselected genes from which we build trees.
ii. Cell type classification only
Spapros optimizes for two objectives:
cell type identification
recovery of transcriptomic variation
The procedure prioritzes the 1st objective over the 2nd. The variation recovery also leads to the selection of genes that show gradients in multiple cell types (see the example of FOS in our preprint, Fig. 3). In classical gene panel selection approaches the focus lies on marker genes, ideally markers that are specific to single cell types. In the downstream analysis of spatial data genes like FOS are not useful for identifying the major cell types, these
genes should even be excluded from the clustering to get better separated cell type clusters. If you don’t want to select genes like FOS and only focus on the first objective you can skip the PCA-based selection by setting n_pca_genes=0. In that case only trees on DE genes are trained, the PCA related steps are neglected.
[9]:
# create an instance of the ProbesetSelector class
selector = sp.se.ProbesetSelector(
adata,
n=30,
n_pca_genes=0,
celltype_key="celltype",
verbosity=1,
save_dir=None,
)
Note: No PCA selection will be performed since n_pca_genes = 0. The selected genes will only be based on the DE forests. In that case it can happen that fewer than n = 30 genes are selected. To get n = 30 exclude the selected genes of the first run from adata, rerun the method, and combine the results of the two runs.
Note: The following celltypes' test set sizes for forest training are below min_test_n (=20):
Dendritic cells : 9
Megakaryocytes : 3
The genes selected for those cell types potentially don't generalize well. Find the genes for each of those cell types in self.genes_of_primary_trees after running self.select_probeset().
[10]:
# select probe set
selector.select_probeset()
[11]:
display(selector.probeset[selector.probeset["selection"]].head(3))
display(selector.probeset[selector.probeset["selection"]].tail(3))
| gene_nr | selection | rank | marker_rank | tree_rank | importance_score | pca_score | pre_selected | prior_selected | pca_selected | celltypes_DE_1vsall | celltypes_DE_specific | celltypes_DE | celltypes_marker | list_only_ct_marker | required_marker | required_list_marker | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PF4 | 1 | True | 1.0 | 1.0 | 1.0 | 1.000000 | 0 | False | False | False | Megakaryocytes | Megakaryocytes | Megakaryocytes | False | True | False | |
| CD79A | 2 | True | 1.0 | 1.0 | 1.0 | 0.914911 | 0 | False | False | False | B cells | B cells | B cells | False | True | False | |
| S100A8 | 3 | True | 1.0 | 1.0 | 1.0 | 0.831315 | 0 | False | False | False | CD14+ Monocytes | CD14+ Monocytes | CD14+ Monocytes | False | True | False |
| gene_nr | selection | rank | marker_rank | tree_rank | importance_score | pca_score | pre_selected | prior_selected | pca_selected | celltypes_DE_1vsall | celltypes_DE_specific | celltypes_DE | celltypes_marker | list_only_ct_marker | required_marker | required_list_marker | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FGFBP2 | 28 | True | 1.0 | NaN | 1.0 | 0.009360 | 0 | False | False | False | NK cells | NK cells | NK cells | False | False | False | |
| HLA-DQB1 | 29 | True | 1.0 | NaN | 1.0 | 0.008832 | 0 | False | False | False | Dendritic cells | Dendritic cells | Dendritic cells | False | False | False | |
| CTSW | 30 | True | 1.0 | NaN | 1.0 | 0.008201 | 0 | False | False | False | CD8 T cells | CD8 T cells | CD8 T cells | False | False | False |
iii. Selection for high numbers of genes (> 150)
Spapros works well over different ranges of numbers of genes to select. However, for higher numbers of genes (~ > 150, can be lower/higher for lower/higher nr of cell types) a dual selection can make sense. This is e.g. relevant for protocols like MERFISH, and also if you want to increase the robustness on the cell type classification objective (less focus on recovery of variation that is independent of cell type labels).
The idea is to run a selection first (we do not fix n and we do not use PCA selected genes for that run), exclude the genes of that selection, run a second (standard) selection, and combine the results of both runs.
[12]:
# First run
# create an instance of the ProbesetSelector class
selector = sp.se.ProbesetSelector(
adata,
n=None,
n_pca_genes=0,
celltype_key="celltype",
verbosity=1,
save_dir=None,
)
# select probe set
selector.select_probeset()
Note: The following celltypes' test set sizes for forest training are below min_test_n (=20):
Dendritic cells : 9
Megakaryocytes : 3
The genes selected for those cell types potentially don't generalize well. Find the genes for each of those cell types in self.genes_of_primary_trees after running self.select_probeset().
[13]:
# Save genes of the first selection
first_selection = selector.probeset[selector.probeset["selection"]].index.tolist()
n_genes_first_selection = len(first_selection)
print(n_genes_first_selection)
33
As the number of cell types is quite low in our example dataset the first selection only lead to 33 genes. Next we select the rest of genes to get to 150.
[14]:
# Second run
# create an instance of the ProbesetSelector class
selector2 = sp.se.ProbesetSelector(
adata[:,[v for v in adata.var_names if v not in first_selection]],
n=150 - n_genes_first_selection,
n_pca_genes=100,
celltype_key="celltype",
verbosity=1,
save_dir=None,
)
# select probe set
selector2.select_probeset()
Note: The following celltypes' test set sizes for forest training are below min_test_n (=20):
Dendritic cells : 9
Megakaryocytes : 3
The genes selected for those cell types potentially don't generalize well. Find the genes for each of those cell types in self.genes_of_primary_trees after running self.select_probeset().
[15]:
second_selection = selector2.probeset[selector2.probeset["selection"]].index.tolist()
final_selection = first_selection + second_selection
print(len(np.unique(final_selection)))
150
This adds up to a final gene set of 150 genes.
For even higher numbers of genes you might want to consider running multiple additional pre selections with n_pca_genes=0.
Side note: unfortunately our plotting functions are only accessible with each individual selector and not the combination of multiple selectors.
iv. Selection with curated marker list
Often you already have some curated list of markers. Such a list can be added as prior knowledge to achieve the following aims
make sure that the signal of the markers in the list are captured by the selected probe set otherwise add markers from the list
add markers for cell types that don’t occur in
adata.obs[celltype_key]
The criteria how markers are added or how the “signal is captured” is described in the following. Note however that if you just want to add certain genes you can also add them with the preselected_genes (see i.) parameter.
Let’s group the cell types in the marker_list into:
ct_group1: cell types that also occur in
adata.obs[celltype_key]ct_group2: cell types that do not occur in
adata.obs[celltype_key]
The key parameters of the ProbesetSelector that define how marker signals are checked for and if markers should be added are:
n_list_markersmarker_corr_th
You can set n_list_markers to an int, then the method will take care to have captured at least n_list_markers per cell type. Note however that the ct_group1 cell types will also already be captured by the genes of the trained trees. Therefore it can make sense to have different numbers for the two groups, which is achieved by setting e.g. n_list_markers = {'adata_celltypes':2, 'list_celltypes':3}. As mentioned above the markers for ct_group1 and ct_group2 are handeled
differently. For ct_group1 the method checks if it already selected n_list_markers that have correlations above marker_corr_th (default: 0.5) with at least n_list_markers for a given cell type, otherwise markers from the list are added. For ct_group2 the correlation criteria doesn’t make much sense since the correlation is calculated on the given cell types in adata and those shouldn’t express a marker for a cell type of ct_group2. Therefore from ct_group2
n_list_markers markers are just added.
Note that there is also the n_min_markers parameter. This one makes sure that enough genes either from DE tests during the selection or (if given) from the marker_list are selected per cell type of ct_group1. So n_min_markers also applies if no marker_list is given.
Let’s define an example marker list. We added 'cell type A' as a ct_group2 (i.e. not in adata.obs[celltype_key]) cell type example.
[16]:
marker_list = {
'CD4 T cells': ['IL7R'],
'CD14+ Monocytes': ['CD14', 'LYZ'],
'B cells': ['MS4A1'],
'CD8 T cells': ['CD8A'],
'NK cells': ['GNLY', 'NKG7'],
'FCGR3A+ Monocytes': ['FCGR3A', 'MS4A7'],
'Dendritic cells': ['FCER1A', 'CST3'],
'Megakaryocytes': ['NAPA-AS1', 'PPBP'],
'cell type A': ["Gene X", "Gene Y", "CAMK2N1"],
}
[17]:
# create an instance of the ProbesetSelector class
selector = sp.se.ProbesetSelector(
adata,
n=None,
celltype_key="celltype",
verbosity=1,
save_dir=None,
marker_list=marker_list,
n_list_markers={'adata_celltypes': 2, 'list_celltypes': 3},
)
# select probe set
selector.select_probeset()
Note: The following celltypes' test set sizes for forest training are below min_test_n (=20):
Dendritic cells : 9
Megakaryocytes : 3
The genes selected for those cell types potentially don't generalize well. Find the genes for each of those cell types in self.genes_of_primary_trees after running self.select_probeset().
[18]:
selector.probeset[selector.probeset["selection"]]
[18]:
| gene_nr | selection | rank | marker_rank | tree_rank | importance_score | pca_score | pre_selected | prior_selected | pca_selected | celltypes_DE_1vsall | celltypes_DE_specific | celltypes_DE | celltypes_marker | list_only_ct_marker | required_marker | required_list_marker | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FCGR3A | 1 | True | 1.0 | 1.0 | 1.0 | 0.612390 | 1.321373 | False | False | True | FCGR3A+ Monocytes | FCGR3A+ Monocytes | FCGR3A+ Monocytes,FCGR3A+ Monocytes | False | True | False | |
| NKG7 | 2 | True | 1.0 | 1.0 | 1.0 | 0.248463 | 1.658470 | False | False | True | NK cells,CD8 T cells | NK cells,CD8 T cells | NK cells,CD8 T cells,NK cells | False | True | False | |
| CST3 | 3 | True | 1.0 | 1.0 | 1.0 | 0.056431 | 1.557674 | False | False | True | CD14+ Monocytes,Dendritic cells | CD14+ Monocytes,Dendritic cells | CD14+ Monocytes,Dendritic cells,Dendritic cells | False | True | False | |
| MS4A1 | 4 | True | 1.0 | 1.0 | 1.0 | 0.012243 | 0.814808 | False | False | True | B cells | False | True | False | |||
| HLA-DPB1 | 5 | True | 1.0 | 2.0 | 1.0 | 0.649235 | 1.997253 | False | False | True | B cells,Dendritic cells | B cells,Dendritic cells | B cells,Dendritic cells | False | True | False | |
| CCL5 | 6 | True | 1.0 | 2.0 | 1.0 | 0.478418 | 2.553322 | False | False | True | CD8 T cells | CD8 T cells | CD8 T cells | False | True | False | |
| IL32 | 7 | True | 1.0 | 2.0 | 1.0 | 0.185111 | 3.377428 | False | False | True | CD4 T cells | CD4 T cells | CD4 T cells | False | True | False | |
| GNLY | 8 | True | 1.0 | 2.0 | 1.0 | 0.075589 | 2.167588 | False | False | True | NK cells | NK cells | NK cells,NK cells | False | True | False | |
| FCER1A | 9 | True | 1.0 | 2.0 | 1.0 | 0.018118 | 0.298179 | False | False | False | Dendritic cells | Dendritic cells | Dendritic cells,Dendritic cells | False | True | False | |
| PF4 | 10 | True | 1.0 | NaN | 1.0 | 1.000000 | 0.810399 | False | False | True | Megakaryocytes | Megakaryocytes | Megakaryocytes | False | False | False | |
| HLA-DQA1 | 11 | True | 1.0 | NaN | 1.0 | 0.562045 | 0.755408 | False | False | True | Dendritic cells | Dendritic cells | Dendritic cells | False | False | False | |
| GZMB | 12 | True | 1.0 | NaN | 1.0 | 0.512924 | 1.252898 | False | False | True | NK cells | NK cells | NK cells | False | False | False | |
| AIF1 | 13 | True | 1.0 | NaN | 1.0 | 0.238008 | 1.527263 | False | False | True | FCGR3A+ Monocytes | FCGR3A+ Monocytes | FCGR3A+ Monocytes | False | False | False | |
| CD79A | 14 | True | 1.0 | NaN | 1.0 | 0.222419 | 1.226536 | False | False | True | B cells | B cells | B cells | False | False | False | |
| S100A8 | 15 | True | 1.0 | NaN | 1.0 | 0.203852 | 2.254612 | False | False | True | CD14+ Monocytes | CD14+ Monocytes | CD14+ Monocytes | False | False | False | |
| HLA-DMA | 16 | True | 1.0 | NaN | 1.0 | 0.167341 | 0.694292 | False | False | True | False | False | False | ||||
| LTB | 17 | True | 1.0 | NaN | 1.0 | 0.139606 | 3.029459 | False | False | True | CD4 T cells | CD4 T cells | CD4 T cells | False | False | False | |
| GZMK | 18 | True | 1.0 | NaN | 1.0 | 0.124733 | 1.680233 | False | False | True | CD8 T cells | CD8 T cells | CD8 T cells | False | False | False | |
| HLA-DRB1 | 19 | True | 1.0 | NaN | 1.0 | 0.097574 | 1.538738 | False | False | True | False | False | False | ||||
| TYROBP | 20 | True | 1.0 | NaN | 1.0 | 0.093040 | 1.352422 | False | False | True | CD14+ Monocytes | CD14+ Monocytes | CD14+ Monocytes | False | False | False | |
| FCN1 | 21 | True | 1.0 | NaN | 1.0 | 0.088904 | 1.070598 | False | False | True | CD14+ Monocytes | CD14+ Monocytes | CD14+ Monocytes | False | False | False | |
| S100A11 | 22 | True | 1.0 | NaN | 1.0 | 0.078515 | 2.468161 | False | False | True | False | False | False | ||||
| GZMH | 23 | True | 1.0 | NaN | 1.0 | 0.058625 | 1.264788 | False | False | True | False | False | False | ||||
| RBM39 | 24 | True | 1.0 | NaN | 1.0 | 0.036062 | 0.657891 | False | False | True | False | False | False | ||||
| HLA-DPA1 | 25 | True | 1.0 | NaN | 1.0 | 0.024234 | 1.429546 | False | False | True | Dendritic cells | Dendritic cells | Dendritic cells | False | False | False | |
| LST1 | 26 | True | 1.0 | NaN | 1.0 | 0.023708 | 1.263540 | False | False | True | FCGR3A+ Monocytes | FCGR3A+ Monocytes | FCGR3A+ Monocytes | False | False | False | |
| LGALS2 | 27 | True | 1.0 | NaN | 1.0 | 0.023468 | 1.126618 | False | False | True | CD14+ Monocytes | CD14+ Monocytes | CD14+ Monocytes | False | False | False | |
| TRAF3IP3 | 28 | True | 1.0 | NaN | 1.0 | 0.011468 | 2.615410 | False | False | True | False | False | False | ||||
| PRF1 | 29 | True | 1.0 | NaN | 1.0 | 0.010033 | 0.807865 | False | False | True | NK cells | NK cells | NK cells | False | False | False | |
| LGALS1 | 30 | True | 1.0 | NaN | 1.0 | 0.009938 | 2.895099 | False | False | True | False | False | False | ||||
| PRDX1 | 31 | True | 1.0 | NaN | 1.0 | 0.009259 | 0.625947 | False | False | True | False | False | False | ||||
| ATP5O | 32 | True | 1.0 | NaN | 1.0 | 0.007816 | 2.455435 | False | False | True | False | False | False | ||||
| C1orf162 | 33 | True | 1.0 | NaN | 1.0 | 0.007754 | 0.889644 | False | False | True | False | False | False | ||||
| CTSS | 34 | True | 1.0 | NaN | 1.0 | 0.007547 | 2.285013 | False | False | True | CD14+ Monocytes | CD14+ Monocytes | CD14+ Monocytes | False | False | False | |
| SELL | 35 | True | 1.0 | NaN | 1.0 | 0.007499 | 2.800353 | False | False | True | False | False | False | ||||
| CTSW | 36 | True | 1.0 | NaN | 1.0 | 0.006245 | 1.300789 | False | False | True | CD8 T cells | CD8 T cells | CD8 T cells | False | False | False | |
| GIMAP4 | 37 | True | 1.0 | NaN | 1.0 | 0.005317 | 2.422619 | False | False | True | False | False | False | ||||
| LINC00926 | 38 | True | 1.0 | NaN | 1.0 | 0.004536 | 0.563208 | False | False | True | False | False | False | ||||
| MS4A6A | 39 | True | 1.0 | NaN | 1.0 | 0.004139 | 0.481730 | False | False | True | False | False | False | ||||
| GSTP1 | 40 | True | 1.0 | NaN | 1.0 | 0.003561 | 1.483085 | False | False | True | False | False | False | ||||
| PDIA3 | 41 | True | 1.0 | NaN | 1.0 | 0.003147 | 0.749059 | False | False | True | False | False | False | ||||
| HOPX | 42 | True | 1.0 | NaN | 1.0 | 0.003119 | 0.910448 | False | False | True | False | False | False | ||||
| ERH | 43 | True | 1.0 | NaN | 1.0 | 0.002811 | 0.586232 | False | False | True | False | False | False | ||||
| COTL1 | 44 | True | 1.0 | NaN | 1.0 | 0.000730 | 2.993346 | False | False | True | FCGR3A+ Monocytes | FCGR3A+ Monocytes | FCGR3A+ Monocytes | False | False | False | |
| CAMK2N1 | 45 | True | 2.0 | 1.0 | NaN | NaN | 0.029484 | False | False | False | cell type A | True | True | False | |||
| Gene X | 46 | True | 2.0 | 2.0 | NaN | NaN | 0.000000 | False | False | False | cell type A | True | True | False | |||
| Gene Y | 47 | True | 2.0 | 3.0 | NaN | NaN | 0.000000 | False | False | False | cell type A | True | True | False | |||
| PPBP | 48 | True | 3.0 | 1.0 | 2.0 | 1.000000 | 1.163331 | False | False | True | Megakaryocytes | Megakaryocytes | Megakaryocytes,Megakaryocytes | False | True | False | |
| CD14 | 49 | True | 3.0 | 1.0 | NaN | NaN | 0.000000 | False | False | False | CD14+ Monocytes | False | True | False | |||
| CD8A | 50 | True | 3.0 | 1.0 | NaN | NaN | 0.000000 | False | False | False | CD8 T cells | False | True | False | |||
| IL7R | 51 | True | 3.0 | 1.0 | NaN | NaN | 0.000000 | False | False | False | CD4 T cells | False | True | False | |||
| NAPA-AS1 | 52 | True | 4.0 | 2.0 | NaN | NaN | 0.035167 | False | False | False | Megakaryocytes | False | True | False | |||
| LYZ | 53 | True | 4.0 | 2.0 | NaN | NaN | 0.000000 | False | False | False | CD14+ Monocytes | False | True | False | |||
| MS4A7 | 54 | True | 4.0 | 2.0 | NaN | NaN | 0.000000 | False | False | False | FCGR3A+ Monocytes | False | True | False |
The required markers that don’t occur in the trees (tree_rank: NaN) are added to fulfill the n_list_markers criteria. Note that the markers for cell type A are ranked higher compared to the markers of the other cell types as those cell types are already captured by the tree_rank=1 genes.
v. Selection with expression constraints
Why epxression constraints?
For different technologies there can be different constraints on the expression levels of genes, e.g. to prevent optical crowding for methods with combinatorial barcoding (MERFISH, ISS, ..).
How expression constraints work
We support expression constraints by setting thresholds for gene expression levels. In practice it is really difficult to set exact thresholds, therefore we introduce smoothed penalty functions that penalize genes based on their expression. I.e. if a gene is just a bit above the threshold but is very important for the selection it will still be selected.
In general you can apply any function to penalize genes or just assign values to genes. The value for each gene needs to be given in adata.obs[penalty_key] and should be in [0,1] (0: maximal penalty, 1: no penalty). During the selection the penalties apply at 3 steps:
PCA-based selection - for each gene the pca score is multipled with the penalty value
DE-based selection - for each gene the DE score is multipled with the penalty value
adding markers from a curated list (
marker_list) - filter genes that have a penalty value below 1 (the threshold can be adjusted viamarker_selection_hparams['penalty_threshold']))
High and low expression constraints
We will construct two constraints: one for too highly expressed genes and one for too lowly expressed genes. We will base them on quantiles of the genes’ expression distributions.
we consider a gene as too highly expressed if at least 1% of cells are above the threshold, i.e.
the 0.99 quantile > upper_thresholdwe consider a gene as too lowly expressed if at least 10% of cells that do express the gene are below the threshold, i.e.
the 0.9 quantile over cells that express the gene < lower_threshold
How to construct the constraints
First you need to find expression level thresholds from experimental experience. Ideally you have spatial measurements of some reference genes that are too highly (or too lowly) expressed (good candidates are abundant genes like MALAT1). Then you can estimate the thresholds based on the expression of those reference genes in a matched scRNA-seq data set. In our example we go with the tresholds 1 and 2 based on log-normalised data.
With the thresholds we construct a smoothed penalty function which needs to be calibrated by testing it on example selections.
[57]:
# Set thresholds
lower_th = 1.0
upper_th = 2
# Calculate quantiles
sp.ut.get_expression_quantile(adata, q=0.99, normalise=False, log1p=False, zeros_to_nan=False)
sp.ut.get_expression_quantile(adata, q=0.9, normalise=False, log1p=False, zeros_to_nan=True)
[69]:
# Calibrate constraints #
# Try different factors (also note that we fixed different base variances 0.1 and 0.5 for lower and upper penalties)
FACTORS = [0.1,1.0,2.0,10]
penalty_fcts = {}
test_selections = {}
for f in FACTORS:
# Get penalty functions for given factor
penalty_fcts[f"lower_{f}"] = sp.ut.plateau_penalty_kernel(var=0.1 * f, x_min=lower_th, x_max=None)
penalty_fcts[f"upper_{f}"] = sp.ut.plateau_penalty_kernel(var=0.5*f, x_min=None, x_max=upper_th)
# Calculate each gene's penalty value
adata.var[f"expr_penalty_lower_{f}"] = penalty_fcts[f"lower_{f}"](adata.var['quantile_0.9 expr > 0'])
adata.var[f"expr_penalty_upper_{f}"] = penalty_fcts[f"upper_{f}"](adata.var['quantile_0.99'])
# PCA and DE selections with penalties
penalty_keys = [f"expr_penalty_lower_{f}",f"expr_penalty_upper_{f}"]
test_selections[f"PCA_{f}"] = sp.se.select_pca_genes(adata, n=100, penalty_keys=penalty_keys, inplace=False)
test_selections[f"DE_{f}"] = sp.se.select_DE_genes(adata, n=100, obs_key="celltype", penalty_keys=penalty_keys, inplace=False)
# Plot selections for different factors
for method in ["DE", "PCA"]:
print(f"### {method} ###")
for f in FACTORS:
used_penalties = {f"lower_{f}": penalty_fcts[f"lower_{f}"], f"upper_{f}": penalty_fcts[f"upper_{f}"]}
sp.pl.selection_histogram(
adata,
{f"{method}_{f}": test_selections[f"{method}_{f}"]},
x_axis_keys={f"lower_{f}": 'quantile_0.9 expr > 0', f"upper_{f}": 'quantile_0.99'},
background_key="all",
penalty_kernels={f"{method}_{f}": used_penalties},
)
### DE ###
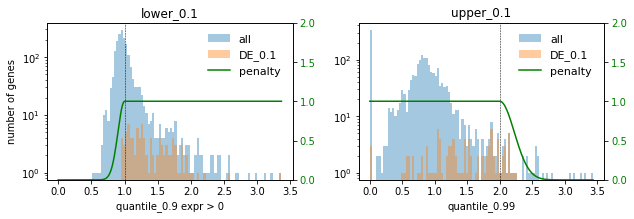
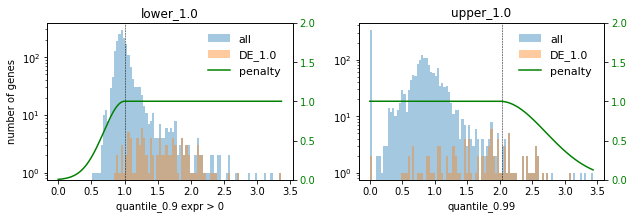
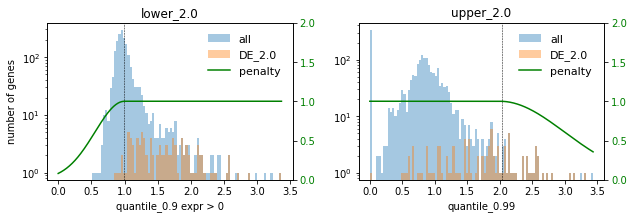
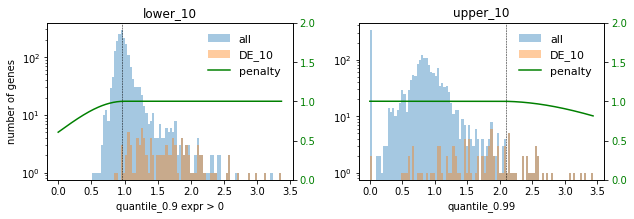
### PCA ###
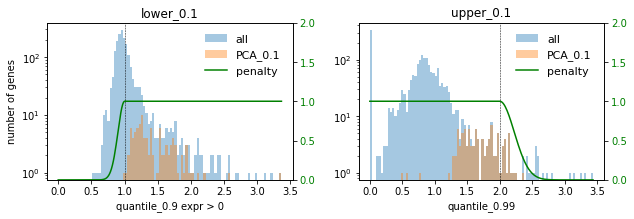
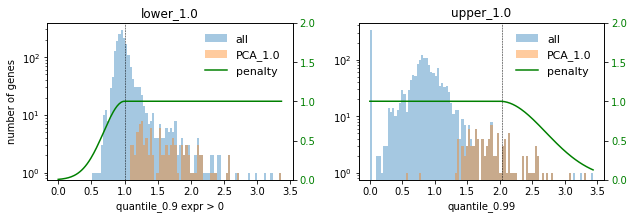
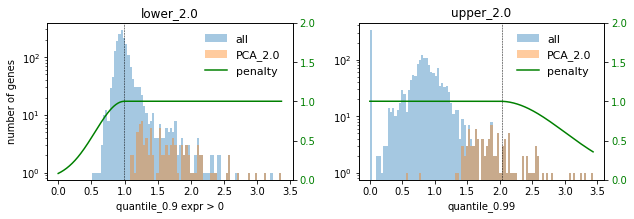
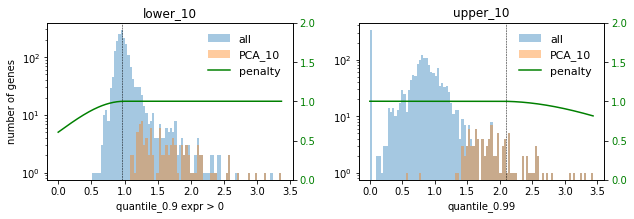
In this setting we choose the factor 0.1 to not select too many highly expressed genes. Lowly expressed genes are not selected anyway for the given selection thresholds.
We apply the expression penalties on multiple selection steps (see below). The “m_penalties<…>” would not be necessary since we don’t provide a marker list. For the case that a marker list is given the penalties should be applied as below: no lower penalty for m_penalties_list_celltypes since we don’t expect cells from that cell type expressing those markers in the given data set.
[70]:
# Run selection with the calibrated expession constraints
FACTOR = 0.1
adata.var["expr_penalty_lower"] = adata.var[f"expr_penalty_lower_{FACTOR}"]
adata.var["expr_penalty_upper"] = adata.var[f"expr_penalty_upper_{FACTOR}"]
# create an instance of the ProbesetSelector class
selector = sp.se.ProbesetSelector(
adata,
n=None,
celltype_key="celltype",
verbosity=1,
save_dir=None,
pca_penalties=["expr_penalty_lower", "expr_penalty_upper"],
DE_penalties=["expr_penalty_lower", "expr_penalty_upper"],
m_penalties_adata_celltypes=["expr_penalty_lower", "expr_penalty_upper"],
m_penalties_list_celltypes=["expr_penalty_upper"],
)
# select probe set
selector.select_probeset()
Note: The following celltypes' test set sizes for forest training are below min_test_n (=20):
Dendritic cells : 9
Megakaryocytes : 3
The genes selected for those cell types potentially don't generalize well. Find the genes for each of those cell types in self.genes_of_primary_trees after running self.select_probeset().
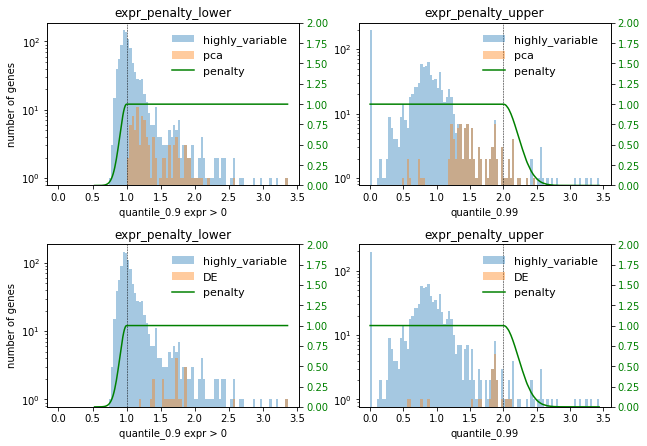
We can then investigate if the genes selected with spapros are within he expression constraints: (the selector.plot_histogram fct probably fails in spapros version 0.1.0, this will be fixed with the next release)
[75]:
selector.plot_histogram(
selections = ["pca", "DE"],
x_axis_keys={"expr_penalty_lower": 'quantile_0.9 expr > 0', "expr_penalty_upper": 'quantile_0.99'},
unapplied_penalty_keys={}
)
vi. Your use case is not captured?
If you face a special use case and you’re unsure if the described schemes would fit to this one, please raise an issue at our github issue section and describe the use case.
3. What’s next?
See our basic evaluation tutorial and advanced evaluation tutorial for detailed instructions to evaluate the selected probesets.
Check out our end to end selection tutorial for combining gene selection and probe design.