End to end selection
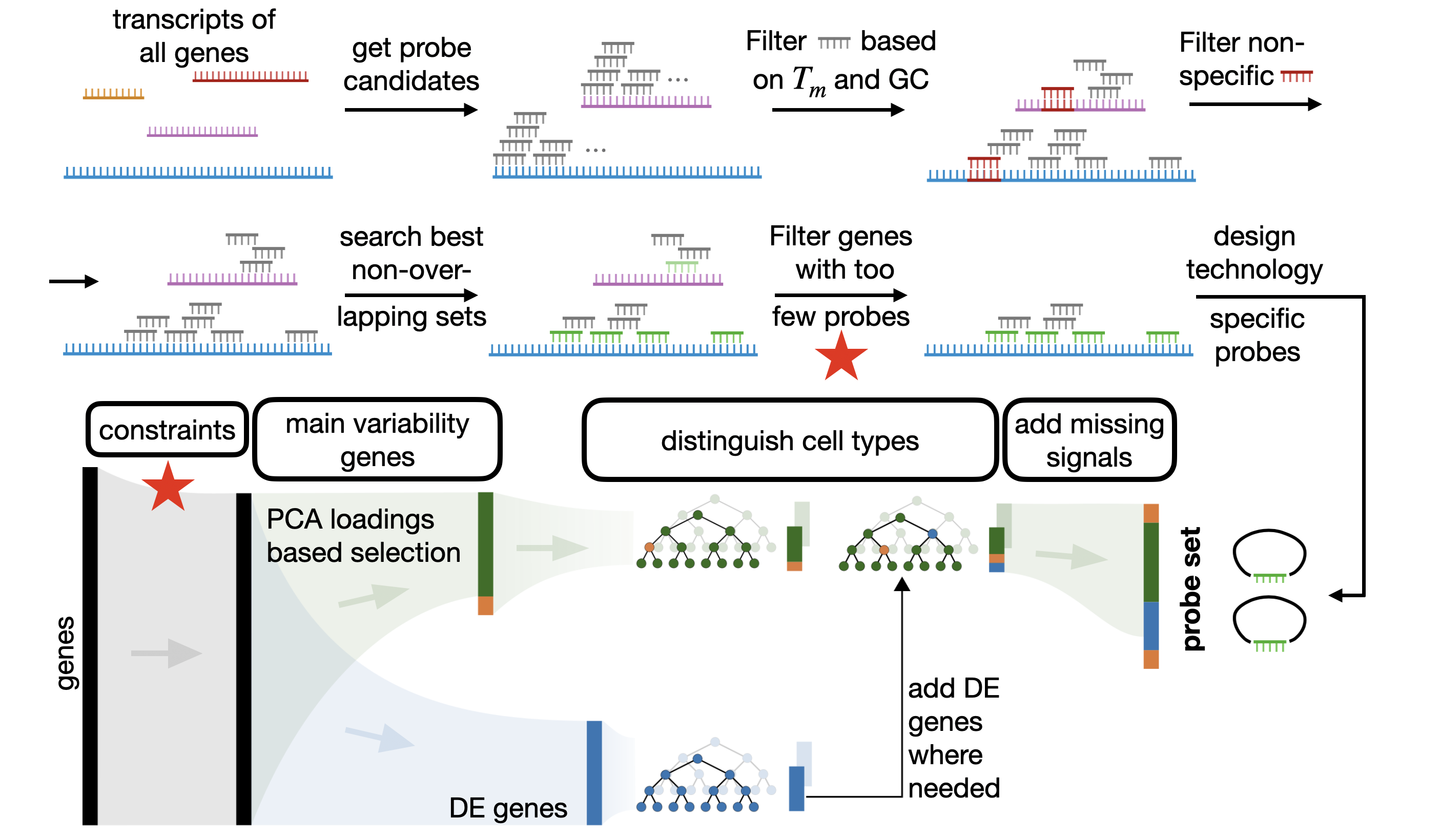
In this tutorial we showcase how to design probesets and select a suitable gene set with the spapros package. For all genes we design probes that fulfill experiment specific requirements and select only genes for which we can design sufficient probes. Spapros then selects genes that can distinguish the cell types in the data set and capture transcriptomic varation beyond cell type labels. The final probe sequences are designed in a last step for all selected genes. The figure below gives and
overview on the pipeline.
[2]:
Import Packages
Besides spapros also install oligo_designer_toolsuite. Therefore, first setup a conda environment (packages is tested for Python 3.9 - 3.10), e.g.:
conda create -n odt python=3.10
conda activate odt
Then, install the required dependencies, i.e. Blast (2.15 or higher), BedTools (2.30 or higher), Bowtie (1.3 or higher) and Bowtie2 (2.5 or higher), that need to be installed independently. To install those tools via conda, please activate the Bioconda and conda-forge channels in your conda environment with and update conda and all packages in your environment:
conda config --add channels conda-forge
conda config --add channels bioconda
conda update --all
conda install "blast>=2.15.0"
conda install "bedtools>=2.30"
conda install "bowtie>=1.3.1"
conda install "bowtie2>=2.5"
All other required packages are automatically installed during the pip installation:
git clone https://github.com/HelmholtzAI-Consultants-Munich/oligo-designer-toolsuite.git
cd oligo-designer-toolsuite
git switch pipelines
pip install -e .
[3]:
import os
import scanpy as sc
sc.settings.verbosity = 0
sc.logging.print_header()
import spapros as sp
print(f"spapros=={sp.__version__}")
from Bio.SeqUtils import MeltingTemp as mt
from oligo_designer_toolsuite.database import OligoDatabase
from oligo_designer_toolsuite.pipelines import GenomicRegionGenerator, ScrinshotProbeDesigner
scanpy==1.10.1 anndata==0.10.7 umap==0.5.6 numpy==1.26.4 scipy==1.13.1 pandas==1.5.3 scikit-learn==1.5.0 statsmodels==0.14.2 igraph==0.11.5 pynndescent==0.5.12
spapros==0.1.5
Load and Preprocess Data
For this tutorial, we use a PBMC example scRNA-seq reference dataset. The count data should be log-normalised and genes should not be scaled to mean=0 and std=1. We can load the processed version of the data, including cell / gene filters, cell type annotations, and the umap embedding, directly with sp.ut.get_processed_pbmc_data() function. For a step by step processing of the PBMC dataset please refer to the basic usage tutorial. For sake
of simplicity, we pre-select the top 1000 highly variable genes for the probe and geneset selection. In real world applications we typically go for top 8000 genes.
[4]:
pbmc_data = sp.ut.get_processed_pbmc_data(n_hvg=1000)
highly_variable_genes = sorted(pbmc_data.var.loc[pbmc_data.var['highly_variable']].index.tolist())
print(f"Number of highly variable genes: {len(highly_variable_genes)}")
Number of highly variable genes: 1000
Probeset Design
Before choosing a gene panel, we design probesets for our given set of 1000 highly variable genes that fulfill certain experiment-specific criteria. Therefore, we first have to create the input fasta files for the probe design pipeline. We can create those files from custom annotation (GFF and fasta) files or download those annotation files directly from the NCBI or Ensembl FTP server. For generating the probe design pipeline input fasta files, we use the GenomicRegionGeneratorclass for
which we need to set the parameters:
dir_output: name of the directory where the output files will be written
[5]:
## Define parameters
dir_output = "output_genomic_region_generator_ncbi"
[6]:
## Setup pipeline
pipeline = GenomicRegionGenerator(dir_output=dir_output)
Generate Genomic Region Files
Our example dataset uses a custom annotation and we create input fasta files for the transcriptome, consisting of exons and exon-exon junctions. Hence, we define custom as source and define the custom-specific parameters listed below. Apart from custom annotation, we can also choose an NCBI or Ensembl annotation. If source=”ncbi” or source=”ensembl” is choosen, the annotation files are automatically downloaded from their servers.
Parameters for annotation loader
source: define annotation source -> currently supported: ncbi, ensembl and custom
Custom annnotation parameters:
source_params:file_annotation: required: GTF file with gene annotationfile_sequence: required: FASTA file with genome sequencefiles_source: optional: original source of the genomic filesspecies: optional: species of provided annotation, leave empty if unknownannotation_release: optional: release number of provided annotation, leave empty if unknowngenome_assembly: optional: genome assembly of provided annotation, leave empty if unknown
For NCBI or Ensembl parameters, see examples in the code below
genomic_regions:gene: create fasta file from gene regions in GFF annotationexon: create fasta file from exon regions in GFF annotation and merge same exons coming from different transcripts into one sequence entry while preserving the transcript informationexon_exon_junction: create fasta file from exon exon junctions in GFF annotation and merge same junctions coming from different transcripts into one sequence entry while preserving the transcript informationcds: create fasta file from cds regions in GFF annotation and merge same cds coming from different transcripts into one sequence entry while preserving the transcript informationintron: create fasta file from regions between exons
exon_exon_junction_block_size: If exon_exon_junction is set to true, specify the block size, i.e. +/- “block_size” bp around the junction. Hint: it does not make sense to set the block size larger than the maximum probe length
Note: if an error occurs for the unzipping of files, this might be due to a faulty download of files from the ftp server. In this case, try to download the files manually from the ftp server and use those files as input for the pipeline with custom input files.
[7]:
## Define parameters for NCBI source
source = "ncbi"
source_params = {
"taxon": "vertebrate_mammalian", # required: taxon of the species, valid taxa are: archaea, bacteria, fungi, invertebrate, mitochondrion, plant, plasmid, plastid, protozoa, vertebrate_mammalian, vertebrate_other, viral
"species": "Homo_sapiens", # required: species name in NCBI download format, e.g. 'Homo_sapiens' for human; see https://ftp.ncbi.nlm.nih.gov/genomes/refseq/ for available species name
"annotation_release": 110 # required: release number of annotation e.g. '109' or '109.20211119' or 'current' to use most recent annotation release. Check out release numbers for NCBI at ftp.ncbi.nlm.nih.gov/refseq/H_sapiens/annotation/annotation_releases/
}
## Define parameters for ensembl source
# source = "ensembl"
# source_params = {
# "species": "homo_sapiens", # required: species name in ensemble download format, e.g. 'homo_sapiens' for human; see http://ftp.ensembl.org/pub/release-108/gtf/ for available species names
# "annotation_release": "109", # required: release number of annotation, e.g. 'release-108' or 'current' to use most recent annotation release. Check out release numbers for ensemble at ftp.ensembl.org/pub/
# }
## Define parameters for custom source
# source = "custom"
# source_params = {
# "file_annotation": "data/custom_GCF_000001405.40_GRCh38.p14_genomic_chr16.gtf",
# "file_sequence": "data/custom_GCF_000001405.40_GRCh38.p14_genomic_chr16.fna",
# "files_source": "NCBI",
# "species": "Homo_sapiens",
# "annotation_release": 110,
# "genome_assembly": "GRCh38",
# }
genomic_regions = {"gene": False, "exon": True, "exon_exon_junction": True, "cds": False, "intron": False}
block_size = 39 # min probe size - 1
[8]:
# Generate the genomic regions
region_generator = pipeline.load_annotations(
source=source,
source_params=source_params,
)
fasta_files = pipeline.generate_genomic_regions(
region_generator=region_generator,
genomic_regions=genomic_regions,
block_size=block_size,
)
2024-07-08 14:35:32,314 [INFO] Parameters Load Annotations:
2024-07-08 14:35:32,316 [INFO] source = ncbi
2024-07-08 14:35:32,317 [INFO] source_params = {'taxon': 'vertebrate_mammalian', 'species': 'Homo_sapiens', 'annotation_release': 110}
2024-07-08 14:44:42,316 [INFO] The following annotation files are used for GTF annotation of regions: /Users/lisa.barros/Desktop/spapros/docs/_tutorials/output_genomic_region_generator_ncbi/annotation/GCF_000001405.40_GRCh38.p14_genomic.gtf and for fasta sequence file: /Users/lisa.barros/Desktop/spapros/docs/_tutorials/output_genomic_region_generator_ncbi/annotation/GCF_000001405.40_GRCh38.p14_genomic.fna .
2024-07-08 14:44:42,381 [INFO] The annotations are from NCBI source, for the species: Homo_sapiens, release number: 110 and genome assembly: GRCh38.p14
2024-07-08 14:48:17,407 [INFO] The genomic region 'exon' was stored in :/Users/lisa.barros/Desktop/spapros/docs/_tutorials/output_genomic_region_generator_ncbi/annotation/exon_annotation_source-NCBI_species-Homo_sapiens_annotation_release-110_genome_assemly-GRCh38.p14.fna.
2024-07-08 15:00:11,519 [INFO] The genomic region 'exon_exon_junction' was stored in :/Users/lisa.barros/Desktop/spapros/docs/_tutorials/output_genomic_region_generator_ncbi/annotation/exon_exon_junction_annotation_source-NCBI_species-Homo_sapiens_annotation_release-110_genome_assemly-GRCh38.p14.fna.
[9]:
fasta_files = ["output_genomic_region_generator_ncbi/annotation/exon_annotation_source-NCBI_species-Homo_sapiens_annotation_release-110_genome_assemly-GRCh38.p14.fna",
"output_genomic_region_generator_ncbi/annotation/exon_exon_junction_annotation_source-NCBI_species-Homo_sapiens_annotation_release-110_genome_assemly-GRCh38.p14.fna"]
Create Probe Database
After generating the input fasta files, we can start with the probe design process. Therefore, we first create an instance of a ProbeDesigner class, where we can choose from ScrinshotProbeDesigner, MerfishProbeDesigner an SeqfishPlusProbeDesigner (see our resource table for an
overview of differences between the technologies). For each of those classes, we need to set the parameter:
gene_ids: a list the genes used to generate the oligos sequences, leave empty list if all the genes should be usedwrite_intermediate_steps: if true, writes the oligo sequences after each step of the pipeline into a tsv filedir_output: name of the directory where the output files will be writtenn_jobs: number of cores used to run the pipeline and number of regions that should be stored in cache. If memory consumption of pipeline is too high reduce this number, if a lot of RAM is available increase this number to decrease runtime
Here, we showcase the probe design of padlock probes for 1000 highly variable genes.
[10]:
## Define parameters
gene_ids = highly_variable_genes # here we use the list of genes of highly variable genes we stored in step 1
write_intermediate_steps = True
dir_output = "output_scrinshot_probe_designer"
n_jobs = 4
[11]:
## Setup pipeline
pipeline = ScrinshotProbeDesigner(
write_intermediate_steps=write_intermediate_steps,
dir_output=dir_output,
n_jobs=n_jobs,
)
After instatiating our ScrinshotProbeDesigner class, we have to create the oligo database. Running the function below, will automatically create all possible probes from the input fasta files for each gene that is provided in the file_regions file.
Parameters for Probe Database
probe_length_min: minimum length of probesprobe_length_max: maximum length of probesprobes_per_gene_min: minimum number of probes that a gene must have before it gets deletedfiles_fasta_oligo_database: fasta file with sequences form which the oligos should be generated. Hint: use the GenomicRegionGenerator class to create fasta files of genomic regions of interest
load_database(). See example code in the cells below (commented).[12]:
## Define parameters
probe_length_min = 40
probe_length_max = 45
min_probes_per_gene = 3 # should be at least "min_probeset_size" probes per gene to create one set
[13]:
## Create initial database
probe_database, file_database = pipeline.create_probe_database(
gene_ids=gene_ids,
probe_length_min=probe_length_min,
probe_length_max=probe_length_max,
files_fasta_oligo_database=fasta_files,
min_probes_per_gene=min_probes_per_gene,
)
2024-07-08 15:00:47,199 [INFO] Parameters Create Database:
2024-07-08 15:00:47,204 [INFO] Function: create_probe_database
2024-07-08 15:00:47,205 [INFO] Parameter: gene_ids = ['AAGAB', 'AATF', 'ABCC10', 'ABHD12', 'ABHD17B', 'ABHD5', 'ABRACL', 'ABT1', 'AC005082.12', 'AC074138.3', 'AC093323.3', 'ACAP1', 'ACBD3', 'ACD', 'ACOT13', 'ACP1', 'ACRBP', 'ACTL6A', 'ACTR6', 'ACVR2A', 'ADAL', 'ADAM10', 'ADAM28', 'ADD1', 'ADIPOR2', 'ADPRM', 'ADSL', 'AEBP1', 'AGPAT1', 'AHSA1', 'AIF1', 'AIM2', 'AKTIP', 'AL928768.3', 'ALKBH7', 'ANAPC13', 'ANKAR', 'ANKEF1', 'ANKRD27', 'ANKRD54', 'AP001462.6', 'AP003419.16', 'AP3M2', 'AP4B1-AS1', 'AP4S1', 'APOBEC3A', 'APOBEC3B', 'APOBEC3G', 'AQP3', 'ARHGAP11A', 'ARHGAP19', 'ARHGAP24', 'ARHGAP33', 'ARHGAP6', 'ARID4A', 'ARIH2OS', 'ARL2', 'ARL2BP', 'ARL4A', 'ARL6IP5', 'ARMC7', 'ARMCX5', 'ARRDC3', 'ARRDC4', 'ARSD', 'ARSG', 'ARVCF', 'ASB8', 'ASXL2', 'ATAD3C', 'ATF7IP2', 'ATG16L1', 'ATP10A', 'ATP5H', 'ATP5O', 'ATP5SL', 'ATP6V0E2', 'ATXN1L', 'ATXN3', 'AURKC', 'BABAM1', 'BACE2', 'BAZ2A', 'BBX', 'BCDIN3D', 'BET1', 'BEX4', 'BGLAP', 'BLNK', 'BLZF1', 'BMPR2', 'BNIP2', 'BOLA1', 'BOLA3', 'BRAT1', 'BRWD1', 'BTN3A1', 'BTN3A2', 'BUB3', 'C10orf32', 'C12orf45', 'C14orf1', 'C14orf166', 'C14orf80', 'C15orf57', 'C16orf13', 'C16orf52', 'C16orf54', 'C16orf58', 'C16orf74', 'C16orf80', 'C17orf59', 'C17orf62', 'C19orf33', 'C19orf52', 'C1QA', 'C1QB', 'C1QC', 'C1orf162', 'C1orf35', 'C21orf33', 'C2CD4D', 'C2orf76', 'C2orf88', 'C3orf18', 'C5orf15', 'C5orf42', 'C8orf44', 'C9orf142', 'C9orf16', 'C9orf37', 'CAMK1D', 'CAMK2G', 'CAMK2N1', 'CAPN12', 'CARHSP1', 'CARS', 'CASC4', 'CBX5', 'CCDC115', 'CCDC122', 'CCDC66', 'CCDC91', 'CCL3', 'CCL4', 'CCL5', 'CCND2', 'CCNG1', 'CCP110', 'CCT4', 'CCT7', 'CD160', 'CD19', 'CD2', 'CD247', 'CD274', 'CD2AP', 'CD320', 'CD72', 'CD79A', 'CD79B', 'CD82', 'CD9', 'CD96', 'CDC123', 'CDC16', 'CDC37', 'CDC40', 'CDK19', 'CDKN2A', 'CEACAM4', 'CEBPB', 'CECR5', 'CEP120', 'CEP68', 'CEP85L', 'CEPT1', 'CES4A', 'CGRRF1', 'CHD2', 'CHD7', 'CHERP', 'CHI3L2', 'CHPF2', 'CIAPIN1', 'CISD1', 'CISH', 'CITED4', 'CKS1B', 'CLDN5', 'CLEC2B', 'CLIC3', 'CLNS1A', 'CLPX', 'CLU', 'CLYBL', 'CMTM5', 'CNEP1R1', 'COMMD10', 'COQ7', 'CORO1B', 'COTL1', 'CPNE2', 'CPQ', 'CPSF3L', 'CR1', 'CRIP3', 'CRTC2', 'CST3', 'CST7', 'CTA-29F11.1', 'CTB-113I20.2', 'CTB-152G17.6', 'CTC-444N24.11', 'CTD-2015H6.3', 'CTD-2302E22.4', 'CTD-2368P22.1', 'CTD-2537I9.12', 'CTSS', 'CTSW', 'CWC15', 'CWC27', 'CXCL10', 'CXCL3', 'CYB5B', 'CYTH2', 'DAGLB', 'DCAF5', 'DDI2', 'DDT', 'DDX1', 'DDX17', 'DDX46', 'DDX56', 'DENND1C', 'DENND2D', 'DENND5B', 'DENND6A', 'DERL1', 'DEXI', 'DHX34', 'DHX9', 'DIDO1', 'DIMT1', 'DIS3', 'DISP1', 'DLST', 'DMTN', 'DNAJA3', 'DNAJB14', 'DNAJC10', 'DNAJC15', 'DNAJC2', 'DNAJC27', 'DNASE1L3', 'DNMT3A', 'DOK3', 'DPH6', 'DPY19L4', 'DRAXIN', 'DSCR3', 'DTX3', 'DUS3L', 'DUSP10', 'EAF2', 'EARS2', 'ECHDC1', 'EDC3', 'EID2', 'EIF1AY', 'EIF1B', 'EIF2B1', 'EIF3D', 'ELANE', 'ELOF1', 'ELOVL4', 'ELP6', 'EMB', 'EMG1', 'EML6', 'ENTPD3-AS1', 'EOGT', 'ERH', 'ERV3-1', 'EVA1B', 'EWSR1', 'EXOC6', 'F5', 'FADS1', 'FAM107B', 'FAM173A', 'FAM210B', 'FAM96A', 'FAM98A', 'FBXL14', 'FBXO21', 'FBXO33', 'FBXO4', 'FBXW4', 'FCER1A', 'FCER1G', 'FCGR2B', 'FCGR3A', 'FCN1', 'FCRLA', 'FEM1A', 'FERMT3', 'FGFBP2', 'FH', 'FHL1', 'FKBP3', 'FKBP5', 'FLOT1', 'FMO4', 'FN3KRP', 'FNBP4', 'FNTA', 'FOPNL', 'FRY-AS1', 'FUS', 'FXN', 'FYB', 'G0S2', 'GADD45B', 'GALT', 'GBGT1', 'GBP1', 'GDF11', 'GFER', 'GGA3', 'GGNBP2', 'GIMAP2', 'GIMAP4', 'GIMAP5', 'GIMAP7', 'GIT2', 'GMPPA', 'GNE', 'GNG11', 'GNG3', 'GNLY', 'GNPAT', 'GOLGB1', 'GP9', 'GPATCH4', 'GPKOW', 'GPR171', 'GPR183', 'GPR35', 'GPS1', 'GPX1', 'GRAP', 'GRN', 'GSTP1', 'GTPBP6', 'GUSB', 'GYS1', 'GZMA', 'GZMB', 'GZMH', 'GZMK', 'HAGH', 'HBA1', 'HBP1', 'HCFC2', 'HDAC1', 'HDAC5', 'HDAC9', 'HELQ', 'HEMK1', 'HERPUD2', 'HIST1H1B', 'HIST1H2AC', 'HIST1H2AH', 'HLA-DMA', 'HLA-DMB', 'HLA-DOB', 'HLA-DPA1', 'HLA-DPB1', 'HLA-DQA1', 'HLA-DQB1', 'HLA-DRB1', 'HMBOX1', 'HMGCL', 'HMGXB4', 'HNRNPH3', 'HOOK2', 'HOPX', 'HSPB11', 'HVCN1', 'ICAM2', 'ICOS', 'ICOSLG', 'ID2', 'IDUA', 'IFFO1', 'IFI27', 'IFIT1', 'IFIT2', 'IFITM3', 'IGFBP7', 'IGJ', 'IGLL5', 'IL1B', 'IL1RAP', 'IL23A', 'IL24', 'IL27RA', 'IL32', 'IL6', 'IL8', 'ILF3', 'ILF3-AS1', 'ING5', 'INSL3', 'INTS12', 'INTS2', 'IP6K1', 'IQCE', 'IRF8', 'IRF9', 'ISCA2', 'ISOC1', 'ITGA2B', 'ITGB7', 'ITM2A', 'ITSN2', 'JAKMIP1', 'JUND', 'KARS', 'KCNG1', 'KCNQ1OT1', 'KIAA0040', 'KIAA0125', 'KIAA0196', 'KIAA1430', 'KIF3A', 'KIF3C', 'KIF5B', 'KLHL24', 'KLRB1', 'KLRG1', 'KRBOX4', 'LAMP3', 'LARS', 'LAT2', 'LBR', 'LDLRAP1', 'LGALS1', 'LGALS2', 'LGALS3', 'LILRA4', 'LIN52', 'LINC00494', 'LINC00662', 'LINC00886', 'LINC00926', 'LINC00936', 'LINC01013', 'LIX1L', 'LONRF1', 'LPIN1', 'LRBA', 'LRRIQ3', 'LSM14A', 'LST1', 'LTB', 'LTV1', 'LUC7L', 'LUC7L3', 'LYAR', 'LYPD2', 'LYPLA1', 'LYRM4', 'LYSMD4', 'LZTS2', 'MADD', 'MAEA', 'MAGEH1', 'MAL', 'MALT1', 'MAP2K7', 'MARCKSL1', 'MCF2L', 'MCM3', 'MDS2', 'MED30', 'MED9', 'METTL21A', 'METTL3', 'METTL8', 'MFF', 'MFSD10', 'MIS18A', 'MKKS', 'MLLT11', 'MLLT6', 'MMADHC', 'MMP9', 'MNAT1', 'MOCS2', 'MORF4L2', 'MPHOSPH10', 'MRM1', 'MRPL1', 'MRPL19', 'MRPL42', 'MRPS12', 'MRPS33', 'MS4A1', 'MS4A6A', 'MTERFD2', 'MTIF2', 'MTRF1', 'MUM1', 'MYADM', 'MYCBP2', 'MYL9', 'MYO1E', 'MYOM2', 'MZB1', 'MZT1', 'NAA20', 'NAP1L4', 'NAPA-AS1', 'NARG2', 'NAT9', 'NBR1', 'NCOR2', 'NCR3', 'NDUFA10', 'NDUFA12', 'NECAB3', 'NEFH', 'NEK8', 'NELFB', 'NEMF', 'NFAT5', 'NFE2L2', 'NFIC', 'NFU1', 'NIT2', 'NKAP', 'NKG7', 'NKTR', 'NME3', 'NME6', 'NMNAT3', 'NNT-AS1', 'NOC4L', 'NOG', 'NOL11', 'NONO', 'NOP58', 'NPC2', 'NPHP3', 'NPRL2', 'NR2C1', 'NR3C1', 'NSA2', 'NT5C', 'NT5C3A', 'NUDCD1', 'NUDT16L1', 'NUP54', 'NXT2', 'OARD1', 'OAT', 'OBSCN', 'ODC1', 'ORAI1', 'ORC2', 'OSBPL1A', 'OSBPL7', 'OXLD1', 'P2RX5', 'P2RY10', 'PACS1', 'PACSIN2', 'PAICS', 'PARP1', 'PARS2', 'PASK', 'PAWR', 'PAXIP1-AS1', 'PBLD', 'PBRM1', 'PCNA', 'PCSK7', 'PDCD1', 'PDCD2L', 'PDE6B', 'PDIA3', 'PDIK1L', 'PDK2', 'PDXDC1', 'PDZD4', 'PEMT', 'PEX16', 'PEX26', 'PF4', 'PGM1', 'PGM2L1', 'PHACTR4', 'PHF12', 'PHF14', 'PHF3', 'PIGF', 'PIGU', 'PIGX', 'PIK3R1', 'PITHD1', 'PITPNA-AS1', 'PJA1', 'PKIG', 'PLA2G12A', 'PLCL1', 'PLD6', 'PLEKHA1', 'PLEKHA3', 'PLRG1', 'PMEPA1', 'PNOC', 'POLR2I', 'POLR2K', 'POLR3E', 'POMT1', 'PPA2', 'PPBP', 'PPIE', 'PPIG', 'PPIL2', 'PPIL4', 'PPP1R14A', 'PPP1R2', 'PPP2R1B', 'PPP6C', 'PPT2-EGFL8', 'PQBP1', 'PRAF2', 'PRDX1', 'PRELID2', 'PRF1', 'PRICKLE1', 'PRKACB', 'PRKCB', 'PRKD2', 'PRMT2', 'PRNP', 'PRPF31', 'PRPS2', 'PRR5', 'PSMD14', 'PTCRA', 'PTGDR', 'PTGDS', 'PTGES2', 'PTPN7', 'PURA', 'PWP1', 'PXMP4', 'PYCARD', 'R3HDM1', 'R3HDM2', 'RAB40C', 'RABEP2', 'RABL6', 'RAD51B', 'RALBP1', 'RALY', 'RASD1', 'RASGRP2', 'RBM25', 'RBM26-AS1', 'RBM39', 'RBM4', 'RBM48', 'RBM5', 'RBM7', 'RBPJ', 'RCE1', 'RCHY1', 'RCL1', 'RCN2', 'RDH14', 'RELB', 'REXO2', 'RFC1', 'RFC5', 'RFNG', 'RFPL2', 'RGS14', 'RIC3', 'RIOK1', 'RIOK2', 'RNF113A', 'RNF125', 'RNF139', 'RNF14', 'RNF168', 'RNF187', 'RNF213', 'RNF25', 'RNF26', 'RORA', 'RP1-28O10.1', 'RP11-1055B8.7', 'RP11-138A9.2', 'RP11-141B14.1', 'RP11-142C4.6', 'RP11-162G10.5', 'RP11-164H13.1', 'RP11-178G16.4', 'RP11-18H21.1', 'RP11-211G3.2', 'RP11-219B17.1', 'RP11-219B4.7', 'RP11-252A24.3', 'RP11-291B21.2', 'RP11-314N13.3', 'RP11-324I22.4', 'RP11-349A22.5', 'RP11-378J18.3', 'RP11-390B4.5', 'RP11-398C13.6', 'RP11-400F19.6', 'RP11-421L21.3', 'RP11-428G5.5', 'RP11-432I5.1', 'RP11-468E2.4', 'RP11-488C13.5', 'RP11-493L12.4', 'RP11-527L4.5', 'RP11-545I5.3', 'RP11-589C21.6', 'RP11-5C23.1', 'RP11-701P16.5', 'RP11-706O15.1', 'RP11-70P17.1', 'RP11-727F15.9', 'RP11-798G7.6', 'RP11-879F14.2', 'RP11-950C14.3', 'RP3-325F22.5', 'RP5-1073O3.7', 'RP5-827C21.4', 'RP5-887A10.1', 'RPH3A', 'RPL39L', 'RPL7L1', 'RPN2', 'RPS6KL1', 'RPUSD2', 'RRAGC', 'RRS1', 'RUNDC1', 'S100A11', 'S100A12', 'S100A8', 'S100B', 'SAFB2', 'SAMD1', 'SAMD3', 'SAMSN1', 'SARDH', 'SARS', 'SAT1', 'SCAI', 'SCAPER', 'SCGB3A1', 'SCPEP1', 'SDCCAG8', 'SDPR', 'SEC61A2', 'SELL', 'SEPT11', 'SERAC1', 'SETD1B', 'SF3B1', 'SF3B5', 'SH3GLB1', 'SH3KBP1', 'SHOC2', 'SHPK-1', 'SIAH2', 'SIRPG', 'SIRT1', 'SIVA1', 'SLA', 'SLBP', 'SLC22A4', 'SLC25A11', 'SLC25A12', 'SLC25A14', 'SLC27A1', 'SLC2A13', 'SLC35A2', 'SLC48A1', 'SLFN5', 'SMARCA4', 'SMARCC2', 'SMC2', 'SMCHD1', 'SMDT1', 'SMIM14', 'SMIM7', 'SNAP47', 'SNHG12', 'SNHG8', 'SNTA1', 'SNX29P2', 'SOX13', 'SPARC', 'SPATA7', 'SPG7', 'SPIB', 'SPIN1', 'SPOCD1', 'SPON2', 'SPSB2', 'SREBF1', 'SRM', 'SRP9', 'SRSF6', 'SSBP1', 'ST3GAL2', 'STAMBP', 'STAU2', 'STK17A', 'STK38', 'STMN1', 'STOML2', 'STUB1', 'STX16', 'STX18', 'SUCLG2', 'SUOX', 'SURF1', 'SURF6', 'SWAP70', 'SYCE1', 'SYP', 'SYVN1', 'TACR2', 'TADA2A', 'TAF10', 'TAF12', 'TAF1D', 'TAL1', 'TALDO1', 'TAPBP', 'TARSL2', 'TASP1', 'TBC1D15', 'TBCK', 'TBXA2R', 'TCEAL4', 'TCEAL8', 'TCL1A', 'TCL1B', 'TCP1', 'TDG', 'TERF2IP', 'TGFBRAP1', 'THAP2', 'THEM4', 'THOC7', 'THUMPD3', 'THYN1', 'TIGIT', 'TIMM10B', 'TMEM116', 'TMEM138', 'TMEM140', 'TMEM14B', 'TMEM165', 'TMEM177', 'TMEM194A', 'TMEM219', 'TMEM242', 'TMEM40', 'TMEM60', 'TMEM80', 'TMEM87A', 'TMEM87B', 'TMEM91', 'TMTC2', 'TMX2', 'TMX3', 'TNFRSF17', 'TNFRSF25', 'TNFRSF4', 'TNFRSF9', 'TNFSF10', 'TOP1MT', 'TOP2B', 'TRABD2A', 'TRAF3IP3', 'TRAPPC12-AS1', 'TRAPPC3', 'TREML1', 'TRIM23', 'TRIP12', 'TRIT1', 'TRMT61A', 'TRPM4', 'TSC22D1', 'TSPAN15', 'TSSC1', 'TTC1', 'TTC14', 'TTC3', 'TTC8', 'TTN-AS1', 'TUBB1', 'TUBG2', 'TYMP', 'TYROBP', 'U2SURP', 'UBA5', 'UBAC2', 'UBE2D2', 'UBE2D4', 'UBE2K', 'UBE2Q1', 'UBE3A', 'UBIAD1', 'UBLCP1', 'UBXN4', 'UCK1', 'UNC45A', 'UQCC1', 'URB2', 'URGCP', 'USP30', 'USP33', 'USP36', 'USP38', 'USP5', 'USP7', 'VAMP5', 'VDAC3', 'VIPR1', 'VPS13A', 'VPS13C', 'VPS25', 'VPS26B', 'VPS28', 'VTI1A', 'VTI1B', 'WARS2', 'WBP2NL', 'WDR55', 'WDR91', 'WDYHV1', 'WNK1', 'WTAP', 'XCL2', 'XPOT', 'XRRA1', 'XXbac-BPG299F13.17', 'YEATS2', 'YES1', 'YPEL2', 'YPEL3', 'YTHDF2', 'ZAP70', 'ZBED5-AS1', 'ZBP1', 'ZC3H15', 'ZCCHC11', 'ZCCHC9', 'ZFAND4', 'ZNF175', 'ZNF232', 'ZNF256', 'ZNF263', 'ZNF276', 'ZNF32', 'ZNF350', 'ZNF436', 'ZNF45', 'ZNF493', 'ZNF503', 'ZNF528', 'ZNF559', 'ZNF561', 'ZNF587B', 'ZNF594', 'ZNF653', 'ZNF682', 'ZNF688', 'ZNF718', 'ZNF747', 'ZNF799', 'ZNF836', 'ZNF92', 'ZRANB3', 'ZSWIM6', 'ZUFSP']
2024-07-08 15:00:47,207 [INFO] Parameter: probe_length_min = 40
2024-07-08 15:00:47,208 [INFO] Parameter: probe_length_max = 45
2024-07-08 15:00:47,209 [INFO] Parameter: files_fasta_oligo_database = ['output_genomic_region_generator_ncbi/annotation/exon_annotation_source-NCBI_species-Homo_sapiens_annotation_release-110_genome_assemly-GRCh38.p14.fna', 'output_genomic_region_generator_ncbi/annotation/exon_exon_junction_annotation_source-NCBI_species-Homo_sapiens_annotation_release-110_genome_assemly-GRCh38.p14.fna']
2024-07-08 15:00:47,210 [INFO] Parameter: min_probes_per_gene = 3
2024-07-08 17:17:26,931 [INFO] Step - Create Database: database contains 26663811 oligos from 887 regions.
2024-07-08 17:17:27,020 [DEBUG] handle_msg[456d1e2d0cc8415e90c2a28419ea6040]({'header': {'date': datetime.datetime(2024, 7, 8, 15, 17, 27, 15000, tzinfo=tzutc()), 'msg_id': 'bffef3a3-1674-47de-8b8f-9180581b259b', 'msg_type': 'comm_msg', 'session': 'f15cb79e-4700-4ef8-aed8-65224aba1be6', 'username': '5bb12fba-c395-4a0a-afbc-31e5784dd153', 'version': '5.2'}, 'msg_id': 'bffef3a3-1674-47de-8b8f-9180581b259b', 'msg_type': 'comm_msg', 'parent_header': {}, 'metadata': {}, 'content': {'comm_id': '456d1e2d0cc8415e90c2a28419ea6040', 'data': {'method': 'update', 'state': {'outputs': [{'output_type': 'display_data', 'data': {'text/plain': ' \x1b[35m100%\x1b[0m \x1b[36mDatabase Loading\x1b[0m \x1b[38;2;114;156;31m━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━\x1b[0m \x1b[32m887/887\x1b[0m \x1b[33m1:12:17\x1b[0m < \x1b[36m0:00:00\x1b[0m\n', 'text/html': '<pre style="white-space:pre;overflow-x:auto;line-height:normal;font-family:Menlo,\'DejaVu Sans Mono\',consolas,\'Courier New\',monospace"> <span style="color: #800080; text-decoration-color: #800080">100%</span> <span style="color: #008080; text-decoration-color: #008080">Database Loading</span> <span style="color: #729c1f; text-decoration-color: #729c1f">━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━</span> <span style="color: #008000; text-decoration-color: #008000">887/887</span> <span style="color: #808000; text-decoration-color: #808000">1:12:17</span> < <span style="color: #008080; text-decoration-color: #008080">0:00:00</span>\n</pre>\n'}, 'metadata': {}}]}, 'buffer_paths': []}}, 'buffers': []})
Refine Database with Property Filters
In order to create experiment-specific probes, we have to apply several filter to each probe, e.g. melting temperature or GC content filters.
Parameters for Probe Sequences:
probe_GC_content_min: minimum GC content of oligosprobe_GC_content_max: maximum GC content of oligosprobe_Tm_min: minimum melting temperature of oligosprobe_Tm_max: maximum melting temperature of oligoshomopolymeric_base_n: minimum number of nucleotides to consider it a homopolymeric run per base and all probes with such a homopolymeric run are filtered out, e.g. if A: 5 is set then the sequence “CTGGTAAAAACTGGA” is filtered out, but the sequence “CTGGTAAAACTGGA” is kept
Parameters for Padlock Arms:
arm_Tm_dif_max: maximum melting temperature difference of both arms (difference shouldn’t be higher than 5! But range is not super important, the lower the better)arm_length_min: minimum length of each armarm_Tm_min: minimum melting temperature of each armarm_Tm_max: maximum melting temperature of each arm
Parameters for Detection Oligos:
min_thymines: minimal number of Thymines in detection oligo.detect_oligo_length_min: minimum length of detection probedetect_oligo_length_max: maximum length of detection probe
Note: The melting temperature is used in 2 different stages (probe and detection oligo design), where a few parameters are shared and the others differ. Parameters for melting temperature -> for more information on parameters, see:here
[16]:
## Define parameters
# probe sequence
probe_GC_content_min = 40
probe_GC_content_max = 60
probe_Tm_min = 65
probe_Tm_max = 75
homopolymeric_base_n = {"A": 5, "T": 5, "C": 5, "G": 5}
# padlock arms
arm_Tm_dif_max = 2
arm_length_min = 10
arm_Tm_min = 50
arm_Tm_max = 60
# detection oligos
min_thymines = 2
U_distance = 5
detect_oligo_length_min = 15
detect_oligo_length_max = 40
detect_oligo_Tm_opt = 56
Tm_parameters_probe = {
"check": True, # default
"strict": True, # default
"c_seq": None, # default
"shift": 0, # default
"nn_table": getattr(mt, "DNA_NN3"), # Allawi & SantaLucia (1997)
"tmm_table": getattr(mt, "DNA_TMM1"), #default
"imm_table": getattr(mt, "DNA_IMM1"), #default
"de_table": getattr(mt, "DNA_DE1"), #default
"dnac1": 50, # [nM]
"dnac2": 0, # [nM]
"selfcomp": False, # default
"saltcorr": 7, # Owczarzy et al. (2008)
"Na": 39, # [mM]
"K": 75, # [mM]
"Tris": 20, # [mM]
"Mg": 10, # [mM]
"dNTPs": 0, # [mM] default
}
Tm_chem_correction_param_probe = {
"DMSO": 0, # default
"fmd": 20,
"DMSOfactor": 0.75, # default
"fmdfactor": 0.65, # default
"fmdmethod": 1, # default
"GC": None, # default
}
Tm_salt_correction_param_probe = None
[17]:
####### Load existing database #######
# dir_database = os.path.join(dir_output, "db_probes/1_db_probes_initial")
# probe_database = OligoDatabase(min_oligos_per_region=min_probes_per_gene, write_regions_with_insufficient_oligos=True, lru_db_max_in_memory=n_jobs*2+2, database_name="db_probes", dir_output=dir_output, n_jobs=n_jobs)
# probe_database.load_database(dir_database=dir_database, region_ids=gene_ids, database_overwrite=True)
## Apply property filters
probe_database, file_database = pipeline.filter_by_property(
oligo_database=probe_database,
probe_GC_content_min=probe_GC_content_min,
probe_GC_content_max=probe_GC_content_max,
probe_Tm_min=probe_Tm_min,
probe_Tm_max=probe_Tm_max,
detect_oligo_length_min=detect_oligo_length_min,
detect_oligo_length_max=detect_oligo_length_max,
min_thymines=min_thymines,
arm_Tm_dif_max=arm_Tm_dif_max,
arm_length_min=arm_length_min,
arm_Tm_min=arm_Tm_min,
arm_Tm_max=arm_Tm_max,
homopolymeric_base_n=homopolymeric_base_n,
Tm_parameters_probe=Tm_parameters_probe,
Tm_chem_correction_param_probe=Tm_chem_correction_param_probe,
Tm_salt_correction_param_probe=Tm_salt_correction_param_probe,
)
2024-07-08 17:41:57,263 [INFO] Parameters Property Filters:
2024-07-08 17:41:57,266 [INFO] Function: filter_by_property
2024-07-08 17:41:57,268 [INFO] Parameter: oligo_database = <oligo_designer_toolsuite.database._oligo_database.OligoDatabase object at 0x10d917550>
2024-07-08 17:41:57,270 [INFO] Parameter: probe_GC_content_min = 40
2024-07-08 17:41:57,272 [INFO] Parameter: probe_GC_content_max = 60
2024-07-08 17:41:57,273 [INFO] Parameter: probe_Tm_min = 65
2024-07-08 17:41:57,274 [INFO] Parameter: probe_Tm_max = 75
2024-07-08 17:41:57,275 [INFO] Parameter: detect_oligo_length_min = 15
2024-07-08 17:41:57,276 [INFO] Parameter: detect_oligo_length_max = 40
2024-07-08 17:41:57,278 [INFO] Parameter: min_thymines = 2
2024-07-08 17:41:57,279 [INFO] Parameter: arm_Tm_dif_max = 2
2024-07-08 17:41:57,279 [INFO] Parameter: arm_length_min = 10
2024-07-08 17:41:57,280 [INFO] Parameter: arm_Tm_min = 50
2024-07-08 17:41:57,282 [INFO] Parameter: arm_Tm_max = 60
2024-07-08 17:41:57,284 [INFO] Parameter: homopolymeric_base_n = {'A': 5, 'T': 5, 'C': 5, 'G': 5}
2024-07-08 17:41:57,286 [INFO] Parameter: Tm_parameters_probe = {'check': True, 'strict': True, 'c_seq': None, 'shift': 0, 'nn_table': {'init': (0, 0), 'init_A/T': (2.3, 4.1), 'init_G/C': (0.1, -2.8), 'init_oneG/C': (0, 0), 'init_allA/T': (0, 0), 'init_5T/A': (0, 0), 'sym': (0, -1.4), 'AA/TT': (-7.9, -22.2), 'AT/TA': (-7.2, -20.4), 'TA/AT': (-7.2, -21.3), 'CA/GT': (-8.5, -22.7), 'GT/CA': (-8.4, -22.4), 'CT/GA': (-7.8, -21.0), 'GA/CT': (-8.2, -22.2), 'CG/GC': (-10.6, -27.2), 'GC/CG': (-9.8, -24.4), 'GG/CC': (-8.0, -19.9)}, 'tmm_table': {'AA/TA': (-3.1, -7.8), 'TA/AA': (-2.5, -6.3), 'CA/GA': (-4.3, -10.7), 'GA/CA': (-8.0, -22.5), 'AC/TC': (-0.1, 0.5), 'TC/AC': (-0.7, -1.3), 'CC/GC': (-2.1, -5.1), 'GC/CC': (-3.9, -10.6), 'AG/TG': (-1.1, -2.1), 'TG/AG': (-1.1, -2.7), 'CG/GG': (-3.8, -9.5), 'GG/CG': (-0.7, -19.2), 'AT/TT': (-2.4, -6.5), 'TT/AT': (-3.2, -8.9), 'CT/GT': (-6.1, -16.9), 'GT/CT': (-7.4, -21.2), 'AA/TC': (-1.6, -4.0), 'AC/TA': (-1.8, -3.8), 'CA/GC': (-2.6, -5.9), 'CC/GA': (-2.7, -6.0), 'GA/CC': (-5.0, -13.8), 'GC/CA': (-3.2, -7.1), 'TA/AC': (-2.3, -5.9), 'TC/AA': (-2.7, -7.0), 'AC/TT': (-0.9, -1.7), 'AT/TC': (-2.3, -6.3), 'CC/GT': (-3.2, -8.0), 'CT/GC': (-3.9, -10.6), 'GC/CT': (-4.9, -13.5), 'GT/CC': (-3.0, -7.8), 'TC/AT': (-2.5, -6.3), 'TT/AC': (-0.7, -1.2), 'AA/TG': (-1.9, -4.4), 'AG/TA': (-2.5, -5.9), 'CA/GG': (-3.9, -9.6), 'CG/GA': (-6.0, -15.5), 'GA/CG': (-4.3, -11.1), 'GG/CA': (-4.6, -11.4), 'TA/AG': (-2.0, -4.7), 'TG/AA': (-2.4, -5.8), 'AG/TT': (-3.2, -8.7), 'AT/TG': (-3.5, -9.4), 'CG/GT': (-3.8, -9.0), 'CT/GG': (-6.6, -18.7), 'GG/CT': (-5.7, -15.9), 'GT/CG': (-5.9, -16.1), 'TG/AT': (-3.9, -10.5), 'TT/AG': (-3.6, -9.8)}, 'imm_table': {'AG/TT': (1.0, 0.9), 'AT/TG': (-2.5, -8.3), 'CG/GT': (-4.1, -11.7), 'CT/GG': (-2.8, -8.0), 'GG/CT': (3.3, 10.4), 'GG/TT': (5.8, 16.3), 'GT/CG': (-4.4, -12.3), 'GT/TG': (4.1, 9.5), 'TG/AT': (-0.1, -1.7), 'TG/GT': (-1.4, -6.2), 'TT/AG': (-1.3, -5.3), 'AA/TG': (-0.6, -2.3), 'AG/TA': (-0.7, -2.3), 'CA/GG': (-0.7, -2.3), 'CG/GA': (-4.0, -13.2), 'GA/CG': (-0.6, -1.0), 'GG/CA': (0.5, 3.2), 'TA/AG': (0.7, 0.7), 'TG/AA': (3.0, 7.4), 'AC/TT': (0.7, 0.2), 'AT/TC': (-1.2, -6.2), 'CC/GT': (-0.8, -4.5), 'CT/GC': (-1.5, -6.1), 'GC/CT': (2.3, 5.4), 'GT/CC': (5.2, 13.5), 'TC/AT': (1.2, 0.7), 'TT/AC': (1.0, 0.7), 'AA/TC': (2.3, 4.6), 'AC/TA': (5.3, 14.6), 'CA/GC': (1.9, 3.7), 'CC/GA': (0.6, -0.6), 'GA/CC': (5.2, 14.2), 'GC/CA': (-0.7, -3.8), 'TA/AC': (3.4, 8.0), 'TC/AA': (7.6, 20.2), 'AA/TA': (1.2, 1.7), 'CA/GA': (-0.9, -4.2), 'GA/CA': (-2.9, -9.8), 'TA/AA': (4.7, 12.9), 'AC/TC': (0.0, -4.4), 'CC/GC': (-1.5, -7.2), 'GC/CC': (3.6, 8.9), 'TC/AC': (6.1, 16.4), 'AG/TG': (-3.1, -9.5), 'CG/GG': (-4.9, -15.3), 'GG/CG': (-6.0, -15.8), 'TG/AG': (1.6, 3.6), 'AT/TT': (-2.7, -10.8), 'CT/GT': (-5.0, -15.8), 'GT/CT': (-2.2, -8.4), 'TT/AT': (0.2, -1.5), 'AI/TC': (-8.9, -25.5), 'TI/AC': (-5.9, -17.4), 'AC/TI': (-8.8, -25.4), 'TC/AI': (-4.9, -13.9), 'CI/GC': (-5.4, -13.7), 'GI/CC': (-6.8, -19.1), 'CC/GI': (-8.3, -23.8), 'GC/CI': (-5.0, -12.6), 'AI/TA': (-8.3, -25.0), 'TI/AA': (-3.4, -11.2), 'AA/TI': (-0.7, -2.6), 'TA/AI': (-1.3, -4.6), 'CI/GA': (2.6, 8.9), 'GI/CA': (-7.8, -21.1), 'CA/GI': (-7.0, -20.0), 'GA/CI': (-7.6, -20.2), 'AI/TT': (0.49, -0.7), 'TI/AT': (-6.5, -22.0), 'AT/TI': (-5.6, -18.7), 'TT/AI': (-0.8, -4.3), 'CI/GT': (-1.0, -2.4), 'GI/CT': (-3.5, -10.6), 'CT/GI': (0.1, -1.0), 'GT/CI': (-4.3, -12.1), 'AI/TG': (-4.9, -15.8), 'TI/AG': (-1.9, -8.5), 'AG/TI': (0.1, -1.8), 'TG/AI': (1.0, 1.0), 'CI/GG': (7.1, 21.3), 'GI/CG': (-1.1, -3.2), 'CG/GI': (5.8, 16.9), 'GG/CI': (-7.6, -22.0), 'AI/TI': (-3.3, -11.9), 'TI/AI': (0.1, -2.3), 'CI/GI': (1.3, 3.0), 'GI/CI': (-0.5, -1.3)}, 'de_table': {'AA/.T': (0.2, 2.3), 'AC/.G': (-6.3, -17.1), 'AG/.C': (-3.7, -10.0), 'AT/.A': (-2.9, -7.6), 'CA/.T': (0.6, 3.3), 'CC/.G': (-4.4, -12.6), 'CG/.C': (-4.0, -11.9), 'CT/.A': (-4.1, -13.0), 'GA/.T': (-1.1, -1.6), 'GC/.G': (-5.1, -14.0), 'GG/.C': (-3.9, -10.9), 'GT/.A': (-4.2, -15.0), 'TA/.T': (-6.9, -20.0), 'TC/.G': (-4.0, -10.9), 'TG/.C': (-4.9, -13.8), 'TT/.A': (-0.2, -0.5), '.A/AT': (-0.7, -0.8), '.C/AG': (-2.1, -3.9), '.G/AC': (-5.9, -16.5), '.T/AA': (-0.5, -1.1), '.A/CT': (4.4, 14.9), '.C/CG': (-0.2, -0.1), '.G/CC': (-2.6, -7.4), '.T/CA': (4.7, 14.2), '.A/GT': (-1.6, -3.6), '.C/GG': (-3.9, -11.2), '.G/GC': (-3.2, -10.4), '.T/GA': (-4.1, -13.1), '.A/TT': (2.9, 10.4), '.C/TG': (-4.4, -13.1), '.G/TC': (-5.2, -15.0), '.T/TA': (-3.8, -12.6)}, 'dnac1': 50, 'dnac2': 0, 'selfcomp': False, 'saltcorr': 7, 'Na': 39, 'K': 75, 'Tris': 20, 'Mg': 10, 'dNTPs': 0}
2024-07-08 17:41:57,288 [INFO] Parameter: Tm_chem_correction_param_probe = {'DMSO': 0, 'fmd': 20, 'DMSOfactor': 0.75, 'fmdfactor': 0.65, 'fmdmethod': 1, 'GC': None}
2024-07-08 17:41:57,288 [INFO] Parameter: Tm_salt_correction_param_probe = None
2024-07-08 18:52:48,260 [INFO] Step - Property Filters: database contains 3468382 oligos from 887 regions.
2024-07-08 18:52:48,357 [DEBUG] handle_msg[af295b15415d4d37bd0c05502a7e2966]({'header': {'date': datetime.datetime(2024, 7, 8, 16, 52, 48, 323000, tzinfo=tzutc()), 'msg_id': '94375a4a-c46b-4ea8-aa8f-b1e5092c97ae', 'msg_type': 'comm_msg', 'session': 'f15cb79e-4700-4ef8-aed8-65224aba1be6', 'username': '5bb12fba-c395-4a0a-afbc-31e5784dd153', 'version': '5.2'}, 'msg_id': '94375a4a-c46b-4ea8-aa8f-b1e5092c97ae', 'msg_type': 'comm_msg', 'parent_header': {}, 'metadata': {}, 'content': {'comm_id': 'af295b15415d4d37bd0c05502a7e2966', 'data': {'method': 'update', 'state': {'outputs': [{'output_type': 'display_data', 'data': {'text/plain': ' \x1b[35m100%\x1b[0m \x1b[36mProperty Filter\x1b[0m \x1b[38;2;114;156;31m━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━\x1b[0m \x1b[32m887/887\x1b[0m \x1b[33m0:59:00\x1b[0m < \x1b[36m0:00:00\x1b[0m\n', 'text/html': '<pre style="white-space:pre;overflow-x:auto;line-height:normal;font-family:Menlo,\'DejaVu Sans Mono\',consolas,\'Courier New\',monospace"> <span style="color: #800080; text-decoration-color: #800080">100%</span> <span style="color: #008080; text-decoration-color: #008080">Property Filter</span> <span style="color: #729c1f; text-decoration-color: #729c1f">━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━</span> <span style="color: #008000; text-decoration-color: #008000">887/887</span> <span style="color: #808000; text-decoration-color: #808000">0:59:00</span> < <span style="color: #008080; text-decoration-color: #008080">0:00:00</span>\n</pre>\n'}, 'metadata': {}}]}, 'buffer_paths': []}}, 'buffers': []})
Refine Database with Specificity Filters
In order to create target specific probes, we have to apply several filter to each probe to lower the probability of off-target effects and cross-hybridization.
cross_hybridization_blastn_search_parameters: parameters for the BlastN searchcross_hybridization_blastn_hit_parameters: parameters for the evaluation of BlastN hits
files_fasta_reference_database: fasta file with sequences used as reference for the specificity filters. Multiple fasta files can be used as reference. Hint: use the genomic_region_generator pipeline to create fasta files of genomic regions of interestligation_region_size: size of the seed region around the ligation site for blast seed region filter; set to 0 if ligation region should not be considered for blast searchspecificity_blastn_search_parameters: parameters for the BlastN searchspecificity_blastn_hit_parameters: parameters for the evaluation of BlastN hits
Note: Depending on the number of genes, this step might be time and memory consuming. For high number of genes, you might want to run this step on a bigger machine!
[18]:
## Define parameters
# Cross-hybridization filter
cross_hybridization_blastn_search_parameters = {
"perc_identity": 80,
"strand": "minus", # this parameter is fixed
"word_size": 10,
"dust": "no",
"soft_masking": "false",
"max_target_seqs": 10,
}
cross_hybridization_blastn_hit_parameters = {
"coverage": 80 # can be turned into min_alignment_length
}
# Specificity Filter
files_fasta_reference_database = fasta_files
ligation_region_size = 5
specificity_blastn_search_parameters = {
"perc_identity": 80,
"strand": "minus", # this parameter is fixed, however if reference is whole genome, consider using "both"
"word_size": 10,
"dust": "no",
"soft_masking": "false",
"max_target_seqs": 10,
"max_hsps": 1000,
}
specificity_blastn_hit_parameters = {
"coverage": 50 # can be turned into min_alignment_length if more meaningful
}
[20]:
####### Load existing database #######
# dir_database = os.path.join(dir_output, "db_probes/2_db_probes_property_filter")
# probe_database = OligoDatabase(min_oligos_per_region=min_probes_per_gene, write_regions_with_insufficient_oligos=True, lru_db_max_in_memory=n_jobs*2+2, database_name="db_probes", dir_output=dir_output, n_jobs=n_jobs)
# probe_database.load_database(dir_database=dir_database, region_ids=gene_ids, database_overwrite=True)
## Apply specificity filters
probe_database, file_database = pipeline.filter_by_specificity(
oligo_database=probe_database,
files_fasta_reference_database=files_fasta_reference_database,
specificity_blastn_search_parameters=specificity_blastn_search_parameters,
specificity_blastn_hit_parameters=specificity_blastn_hit_parameters,
cross_hybridization_blastn_search_parameters=cross_hybridization_blastn_search_parameters,
cross_hybridization_blastn_hit_parameters=cross_hybridization_blastn_hit_parameters,
ligation_region_size=ligation_region_size,
arm_Tm_dif_max=arm_Tm_dif_max,
arm_length_min=arm_length_min,
arm_Tm_min=arm_Tm_min,
arm_Tm_max=arm_Tm_max,
Tm_parameters_probe=Tm_parameters_probe,
Tm_chem_correction_param_probe=Tm_chem_correction_param_probe,
Tm_salt_correction_param_probe=Tm_salt_correction_param_probe,
)
2024-07-08 19:46:51,968 [INFO] Parameters Specificity Filters:
2024-07-08 19:46:51,969 [INFO] Function: filter_by_specificity
2024-07-08 19:46:51,972 [INFO] Parameter: oligo_database = <oligo_designer_toolsuite.database._oligo_database.OligoDatabase object at 0x10d917550>
2024-07-08 19:46:51,973 [INFO] Parameter: files_fasta_reference_database = ['output_genomic_region_generator_ncbi/annotation/exon_annotation_source-NCBI_species-Homo_sapiens_annotation_release-110_genome_assemly-GRCh38.p14.fna', 'output_genomic_region_generator_ncbi/annotation/exon_exon_junction_annotation_source-NCBI_species-Homo_sapiens_annotation_release-110_genome_assemly-GRCh38.p14.fna']
2024-07-08 19:46:51,975 [INFO] Parameter: specificity_blastn_search_parameters = {'perc_identity': 80, 'strand': 'minus', 'word_size': 10, 'dust': 'no', 'soft_masking': 'false', 'max_target_seqs': 10, 'max_hsps': 1000}
2024-07-08 19:46:51,977 [INFO] Parameter: specificity_blastn_hit_parameters = {'coverage': 50}
2024-07-08 19:46:51,978 [INFO] Parameter: cross_hybridization_blastn_search_parameters = {'perc_identity': 80, 'strand': 'minus', 'word_size': 10, 'dust': 'no', 'soft_masking': 'false', 'max_target_seqs': 10}
2024-07-08 19:46:51,979 [INFO] Parameter: cross_hybridization_blastn_hit_parameters = {'coverage': 80}
2024-07-08 19:46:51,980 [INFO] Parameter: ligation_region_size = 5
2024-07-08 19:46:51,984 [INFO] Parameter: arm_Tm_dif_max = 2
2024-07-08 19:46:51,986 [INFO] Parameter: arm_length_min = 10
2024-07-08 19:46:51,988 [INFO] Parameter: arm_Tm_min = 50
2024-07-08 19:46:51,990 [INFO] Parameter: arm_Tm_max = 60
2024-07-08 19:46:51,992 [INFO] Parameter: Tm_parameters_probe = {'check': True, 'strict': True, 'c_seq': None, 'shift': 0, 'nn_table': {'init': (0, 0), 'init_A/T': (2.3, 4.1), 'init_G/C': (0.1, -2.8), 'init_oneG/C': (0, 0), 'init_allA/T': (0, 0), 'init_5T/A': (0, 0), 'sym': (0, -1.4), 'AA/TT': (-7.9, -22.2), 'AT/TA': (-7.2, -20.4), 'TA/AT': (-7.2, -21.3), 'CA/GT': (-8.5, -22.7), 'GT/CA': (-8.4, -22.4), 'CT/GA': (-7.8, -21.0), 'GA/CT': (-8.2, -22.2), 'CG/GC': (-10.6, -27.2), 'GC/CG': (-9.8, -24.4), 'GG/CC': (-8.0, -19.9)}, 'tmm_table': {'AA/TA': (-3.1, -7.8), 'TA/AA': (-2.5, -6.3), 'CA/GA': (-4.3, -10.7), 'GA/CA': (-8.0, -22.5), 'AC/TC': (-0.1, 0.5), 'TC/AC': (-0.7, -1.3), 'CC/GC': (-2.1, -5.1), 'GC/CC': (-3.9, -10.6), 'AG/TG': (-1.1, -2.1), 'TG/AG': (-1.1, -2.7), 'CG/GG': (-3.8, -9.5), 'GG/CG': (-0.7, -19.2), 'AT/TT': (-2.4, -6.5), 'TT/AT': (-3.2, -8.9), 'CT/GT': (-6.1, -16.9), 'GT/CT': (-7.4, -21.2), 'AA/TC': (-1.6, -4.0), 'AC/TA': (-1.8, -3.8), 'CA/GC': (-2.6, -5.9), 'CC/GA': (-2.7, -6.0), 'GA/CC': (-5.0, -13.8), 'GC/CA': (-3.2, -7.1), 'TA/AC': (-2.3, -5.9), 'TC/AA': (-2.7, -7.0), 'AC/TT': (-0.9, -1.7), 'AT/TC': (-2.3, -6.3), 'CC/GT': (-3.2, -8.0), 'CT/GC': (-3.9, -10.6), 'GC/CT': (-4.9, -13.5), 'GT/CC': (-3.0, -7.8), 'TC/AT': (-2.5, -6.3), 'TT/AC': (-0.7, -1.2), 'AA/TG': (-1.9, -4.4), 'AG/TA': (-2.5, -5.9), 'CA/GG': (-3.9, -9.6), 'CG/GA': (-6.0, -15.5), 'GA/CG': (-4.3, -11.1), 'GG/CA': (-4.6, -11.4), 'TA/AG': (-2.0, -4.7), 'TG/AA': (-2.4, -5.8), 'AG/TT': (-3.2, -8.7), 'AT/TG': (-3.5, -9.4), 'CG/GT': (-3.8, -9.0), 'CT/GG': (-6.6, -18.7), 'GG/CT': (-5.7, -15.9), 'GT/CG': (-5.9, -16.1), 'TG/AT': (-3.9, -10.5), 'TT/AG': (-3.6, -9.8)}, 'imm_table': {'AG/TT': (1.0, 0.9), 'AT/TG': (-2.5, -8.3), 'CG/GT': (-4.1, -11.7), 'CT/GG': (-2.8, -8.0), 'GG/CT': (3.3, 10.4), 'GG/TT': (5.8, 16.3), 'GT/CG': (-4.4, -12.3), 'GT/TG': (4.1, 9.5), 'TG/AT': (-0.1, -1.7), 'TG/GT': (-1.4, -6.2), 'TT/AG': (-1.3, -5.3), 'AA/TG': (-0.6, -2.3), 'AG/TA': (-0.7, -2.3), 'CA/GG': (-0.7, -2.3), 'CG/GA': (-4.0, -13.2), 'GA/CG': (-0.6, -1.0), 'GG/CA': (0.5, 3.2), 'TA/AG': (0.7, 0.7), 'TG/AA': (3.0, 7.4), 'AC/TT': (0.7, 0.2), 'AT/TC': (-1.2, -6.2), 'CC/GT': (-0.8, -4.5), 'CT/GC': (-1.5, -6.1), 'GC/CT': (2.3, 5.4), 'GT/CC': (5.2, 13.5), 'TC/AT': (1.2, 0.7), 'TT/AC': (1.0, 0.7), 'AA/TC': (2.3, 4.6), 'AC/TA': (5.3, 14.6), 'CA/GC': (1.9, 3.7), 'CC/GA': (0.6, -0.6), 'GA/CC': (5.2, 14.2), 'GC/CA': (-0.7, -3.8), 'TA/AC': (3.4, 8.0), 'TC/AA': (7.6, 20.2), 'AA/TA': (1.2, 1.7), 'CA/GA': (-0.9, -4.2), 'GA/CA': (-2.9, -9.8), 'TA/AA': (4.7, 12.9), 'AC/TC': (0.0, -4.4), 'CC/GC': (-1.5, -7.2), 'GC/CC': (3.6, 8.9), 'TC/AC': (6.1, 16.4), 'AG/TG': (-3.1, -9.5), 'CG/GG': (-4.9, -15.3), 'GG/CG': (-6.0, -15.8), 'TG/AG': (1.6, 3.6), 'AT/TT': (-2.7, -10.8), 'CT/GT': (-5.0, -15.8), 'GT/CT': (-2.2, -8.4), 'TT/AT': (0.2, -1.5), 'AI/TC': (-8.9, -25.5), 'TI/AC': (-5.9, -17.4), 'AC/TI': (-8.8, -25.4), 'TC/AI': (-4.9, -13.9), 'CI/GC': (-5.4, -13.7), 'GI/CC': (-6.8, -19.1), 'CC/GI': (-8.3, -23.8), 'GC/CI': (-5.0, -12.6), 'AI/TA': (-8.3, -25.0), 'TI/AA': (-3.4, -11.2), 'AA/TI': (-0.7, -2.6), 'TA/AI': (-1.3, -4.6), 'CI/GA': (2.6, 8.9), 'GI/CA': (-7.8, -21.1), 'CA/GI': (-7.0, -20.0), 'GA/CI': (-7.6, -20.2), 'AI/TT': (0.49, -0.7), 'TI/AT': (-6.5, -22.0), 'AT/TI': (-5.6, -18.7), 'TT/AI': (-0.8, -4.3), 'CI/GT': (-1.0, -2.4), 'GI/CT': (-3.5, -10.6), 'CT/GI': (0.1, -1.0), 'GT/CI': (-4.3, -12.1), 'AI/TG': (-4.9, -15.8), 'TI/AG': (-1.9, -8.5), 'AG/TI': (0.1, -1.8), 'TG/AI': (1.0, 1.0), 'CI/GG': (7.1, 21.3), 'GI/CG': (-1.1, -3.2), 'CG/GI': (5.8, 16.9), 'GG/CI': (-7.6, -22.0), 'AI/TI': (-3.3, -11.9), 'TI/AI': (0.1, -2.3), 'CI/GI': (1.3, 3.0), 'GI/CI': (-0.5, -1.3)}, 'de_table': {'AA/.T': (0.2, 2.3), 'AC/.G': (-6.3, -17.1), 'AG/.C': (-3.7, -10.0), 'AT/.A': (-2.9, -7.6), 'CA/.T': (0.6, 3.3), 'CC/.G': (-4.4, -12.6), 'CG/.C': (-4.0, -11.9), 'CT/.A': (-4.1, -13.0), 'GA/.T': (-1.1, -1.6), 'GC/.G': (-5.1, -14.0), 'GG/.C': (-3.9, -10.9), 'GT/.A': (-4.2, -15.0), 'TA/.T': (-6.9, -20.0), 'TC/.G': (-4.0, -10.9), 'TG/.C': (-4.9, -13.8), 'TT/.A': (-0.2, -0.5), '.A/AT': (-0.7, -0.8), '.C/AG': (-2.1, -3.9), '.G/AC': (-5.9, -16.5), '.T/AA': (-0.5, -1.1), '.A/CT': (4.4, 14.9), '.C/CG': (-0.2, -0.1), '.G/CC': (-2.6, -7.4), '.T/CA': (4.7, 14.2), '.A/GT': (-1.6, -3.6), '.C/GG': (-3.9, -11.2), '.G/GC': (-3.2, -10.4), '.T/GA': (-4.1, -13.1), '.A/TT': (2.9, 10.4), '.C/TG': (-4.4, -13.1), '.G/TC': (-5.2, -15.0), '.T/TA': (-3.8, -12.6)}, 'dnac1': 50, 'dnac2': 0, 'selfcomp': False, 'saltcorr': 7, 'Na': 39, 'K': 75, 'Tris': 20, 'Mg': 10, 'dNTPs': 0}
2024-07-08 19:46:51,996 [INFO] Parameter: Tm_chem_correction_param_probe = {'DMSO': 0, 'fmd': 20, 'DMSOfactor': 0.75, 'fmdfactor': 0.65, 'fmdmethod': 1, 'GC': None}
2024-07-08 19:46:51,997 [INFO] Parameter: Tm_salt_correction_param_probe = None
2024-07-08 21:42:02,456 [INFO] Step - Specificity Filters: database contains 408735 oligos from 806 regions.
2024-07-08 21:42:02,542 [DEBUG] handle_msg[ecdf5993189c4082b846e0bd4d424bc1]({'header': {'date': datetime.datetime(2024, 7, 8, 19, 42, 2, 520000, tzinfo=tzutc()), 'msg_id': '3d0f9311-1134-4051-a004-bce437d0daed', 'msg_type': 'comm_msg', 'session': 'f15cb79e-4700-4ef8-aed8-65224aba1be6', 'username': '5bb12fba-c395-4a0a-afbc-31e5784dd153', 'version': '5.2'}, 'msg_id': '3d0f9311-1134-4051-a004-bce437d0daed', 'msg_type': 'comm_msg', 'parent_header': {}, 'metadata': {}, 'content': {'comm_id': 'ecdf5993189c4082b846e0bd4d424bc1', 'data': {'method': 'update', 'state': {'outputs': [{'output_type': 'display_data', 'data': {'text/plain': ' \x1b[35m100%\x1b[0m \x1b[36mSpecificity Filter: Exact Match\x1b[0m \x1b[38;2;114;156;31m━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━\x1b[0m \x1b[32m887/887\x1b[0m \x1b[33m0:04:30\x1b[0m < \x1b[36m0:00:00\x1b[0m\n', 'text/html': '<pre style="white-space:pre;overflow-x:auto;line-height:normal;font-family:Menlo,\'DejaVu Sans Mono\',consolas,\'Courier New\',monospace"> <span style="color: #800080; text-decoration-color: #800080">100%</span> <span style="color: #008080; text-decoration-color: #008080">Specificity Filter: Exact Match</span> <span style="color: #729c1f; text-decoration-color: #729c1f">━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━</span> <span style="color: #008000; text-decoration-color: #008000">887/887</span> <span style="color: #808000; text-decoration-color: #808000">0:04:30</span> < <span style="color: #008080; text-decoration-color: #008080">0:00:00</span>\n</pre>\n'}, 'metadata': {}}]}, 'buffer_paths': []}}, 'buffers': []})
2024-07-08 21:42:02,547 [DEBUG] handle_msg[f78d8fb19a4845f5bb559e02ee379443]({'header': {'date': datetime.datetime(2024, 7, 8, 19, 42, 2, 521000, tzinfo=tzutc()), 'msg_id': '00a4381b-1e14-42e5-a94a-246c14c0bee4', 'msg_type': 'comm_msg', 'session': 'f15cb79e-4700-4ef8-aed8-65224aba1be6', 'username': '5bb12fba-c395-4a0a-afbc-31e5784dd153', 'version': '5.2'}, 'msg_id': '00a4381b-1e14-42e5-a94a-246c14c0bee4', 'msg_type': 'comm_msg', 'parent_header': {}, 'metadata': {}, 'content': {'comm_id': 'f78d8fb19a4845f5bb559e02ee379443', 'data': {'method': 'update', 'state': {'outputs': [{'output_type': 'display_data', 'data': {'text/plain': ' \x1b[35m100%\x1b[0m \x1b[36mSpecificity Filter: Blastn Specificity\x1b[0m \x1b[38;2;114;156;31m━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━\x1b[0m \x1b[32m887/887\x1b[0m \x1b[33m1:08:54\x1b[0m < \x1b[36m0:00:00\x1b[0m\n', 'text/html': '<pre style="white-space:pre;overflow-x:auto;line-height:normal;font-family:Menlo,\'DejaVu Sans Mono\',consolas,\'Courier New\',monospace"> <span style="color: #800080; text-decoration-color: #800080">100%</span> <span style="color: #008080; text-decoration-color: #008080">Specificity Filter: Blastn Specificity</span> <span style="color: #729c1f; text-decoration-color: #729c1f">━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━</span> <span style="color: #008000; text-decoration-color: #008000">887/887</span> <span style="color: #808000; text-decoration-color: #808000">1:08:54</span> < <span style="color: #008080; text-decoration-color: #008080">0:00:00</span>\n</pre>\n'}, 'metadata': {}}]}, 'buffer_paths': []}}, 'buffers': []})
2024-07-08 21:42:02,552 [DEBUG] handle_msg[086807ee363b4bbea301fac1c93c5444]({'header': {'date': datetime.datetime(2024, 7, 8, 19, 42, 2, 521000, tzinfo=tzutc()), 'msg_id': '4d438cb6-e1cd-4b07-9718-cb0949ab2096', 'msg_type': 'comm_msg', 'session': 'f15cb79e-4700-4ef8-aed8-65224aba1be6', 'username': '5bb12fba-c395-4a0a-afbc-31e5784dd153', 'version': '5.2'}, 'msg_id': '4d438cb6-e1cd-4b07-9718-cb0949ab2096', 'msg_type': 'comm_msg', 'parent_header': {}, 'metadata': {}, 'content': {'comm_id': '086807ee363b4bbea301fac1c93c5444', 'data': {'method': 'update', 'state': {'outputs': [{'output_type': 'display_data', 'data': {'text/plain': ' \x1b[35m100%\x1b[0m \x1b[36mSpecificity Filter: Blastn Crosshybridization\x1b[0m \x1b[38;2;114;156;31m━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━\x1b[0m \x1b[32m806/806\x1b[0m \x1b[33m0:02:35\x1b[0m < \x1b[36m0:00:00\x1b[0m\n', 'text/html': '<pre style="white-space:pre;overflow-x:auto;line-height:normal;font-family:Menlo,\'DejaVu Sans Mono\',consolas,\'Courier New\',monospace"> <span style="color: #800080; text-decoration-color: #800080">100%</span> <span style="color: #008080; text-decoration-color: #008080">Specificity Filter: Blastn Crosshybridization</span> <span style="color: #729c1f; text-decoration-color: #729c1f">━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━</span> <span style="color: #008000; text-decoration-color: #008000">806/806</span> <span style="color: #808000; text-decoration-color: #808000">0:02:35</span> < <span style="color: #008080; text-decoration-color: #008080">0:00:00</span>\n</pre>\n'}, 'metadata': {}}]}, 'buffer_paths': []}}, 'buffers': []})
Create Probe Sets of Non-Overlapping Probes
After applying different sets of filters to the probe database, we will create probesets for each gene, which are sets of probes that do not overlap and have a high efficiency score (calculated from melting temperature, GC content and isoform consensus).
Parameters for the Oligo Set Selection
probe_isoform_weight: weight of the isoform consensus of the probe in the efficiency scoreprobe_GC_content_opt: max and min values are defiend aboveprobe_GC_weight: weight of the GC content of the probe in the efficiency scoreprobe_Tm_opt: max and min values are defiend aboveprobe_Tm_weight: weight of the Tm of the probe in the efficiency scoreprobeset_size_min: minimum size of probe sets (in case there exist no set of the optimal size) -> genes with less oligos will be filtered out and stored in regions_with_insufficient_oligos_for_db_probesprobeset_size_opt: optimal size of probe setsdistance_between_probes: how much overlap should be allowed between oligos, e.g. if oligos can overlpap x bases choose -x, if oligos can be next to one another choose 0, if oligos should be x bases apart choose xn_sets: maximum number of sets to generatemax_graph_size: maximum number of oligos that are taken into consisderation in the last step (5000 -> ~5GB, 2500 -> ~1GB)
[21]:
## Define parameters
probe_isoform_weight = 2
probe_GC_weight = 1
probe_GC_content_opt = 50
probe_Tm_weight = 1
probe_Tm_opt = 70
probeset_size_min = 3
probeset_size_opt = 5
distance_between_probes = 0
n_sets = 100
max_graph_size = 5000
[23]:
####### Load existing database #######
# dir_database = os.path.join(dir_output, "db_probes/3_db_probes_specificity_filter")
# probe_database = OligoDatabase(min_oligos_per_region=min_probes_per_gene, write_regions_with_insufficient_oligos=True, lru_db_max_in_memory=n_jobs*2+2, database_name="db_probes", dir_output=dir_output, n_jobs=n_jobs)
# probe_database.load_database(dir_database=dir_database, region_ids=gene_ids, database_overwrite=True)
## Apply probe selection
probe_database, file_database, dir_probesets = pipeline.create_probe_sets(
oligo_database=probe_database,
probe_isoform_weight=probe_isoform_weight,
probe_Tm_weight=probe_Tm_weight,
probe_Tm_min=probe_Tm_min,
probe_Tm_opt=probe_Tm_opt,
probe_Tm_max=probe_Tm_max,
Tm_parameters_probe=Tm_parameters_probe,
Tm_chem_correction_param_probe=Tm_chem_correction_param_probe,
Tm_salt_correction_param_probe=Tm_salt_correction_param_probe,
probe_GC_weight=probe_GC_weight,
probe_GC_content_min=probe_GC_content_min,
probe_GC_content_opt=probe_GC_content_opt,
probe_GC_content_max=probe_GC_content_max,
probeset_size_opt=probeset_size_opt,
probeset_size_min=probeset_size_min,
max_graph_size=max_graph_size,
n_sets=n_sets,
distance_between_probes=distance_between_probes,
)
2024-07-09 00:36:23,067 [INFO] Parameters Set Selection:
2024-07-09 00:36:23,070 [INFO] Function: create_probe_sets
2024-07-09 00:36:23,072 [INFO] Parameter: oligo_database = <oligo_designer_toolsuite.database._oligo_database.OligoDatabase object at 0x10d917550>
2024-07-09 00:36:23,075 [INFO] Parameter: probe_isoform_weight = 2
2024-07-09 00:36:23,077 [INFO] Parameter: probe_Tm_weight = 1
2024-07-09 00:36:23,078 [INFO] Parameter: probe_Tm_min = 65
2024-07-09 00:36:23,081 [INFO] Parameter: probe_Tm_opt = 70
2024-07-09 00:36:23,084 [INFO] Parameter: probe_Tm_max = 75
2024-07-09 00:36:23,086 [INFO] Parameter: Tm_parameters_probe = {'check': True, 'strict': True, 'c_seq': None, 'shift': 0, 'nn_table': {'init': (0, 0), 'init_A/T': (2.3, 4.1), 'init_G/C': (0.1, -2.8), 'init_oneG/C': (0, 0), 'init_allA/T': (0, 0), 'init_5T/A': (0, 0), 'sym': (0, -1.4), 'AA/TT': (-7.9, -22.2), 'AT/TA': (-7.2, -20.4), 'TA/AT': (-7.2, -21.3), 'CA/GT': (-8.5, -22.7), 'GT/CA': (-8.4, -22.4), 'CT/GA': (-7.8, -21.0), 'GA/CT': (-8.2, -22.2), 'CG/GC': (-10.6, -27.2), 'GC/CG': (-9.8, -24.4), 'GG/CC': (-8.0, -19.9)}, 'tmm_table': {'AA/TA': (-3.1, -7.8), 'TA/AA': (-2.5, -6.3), 'CA/GA': (-4.3, -10.7), 'GA/CA': (-8.0, -22.5), 'AC/TC': (-0.1, 0.5), 'TC/AC': (-0.7, -1.3), 'CC/GC': (-2.1, -5.1), 'GC/CC': (-3.9, -10.6), 'AG/TG': (-1.1, -2.1), 'TG/AG': (-1.1, -2.7), 'CG/GG': (-3.8, -9.5), 'GG/CG': (-0.7, -19.2), 'AT/TT': (-2.4, -6.5), 'TT/AT': (-3.2, -8.9), 'CT/GT': (-6.1, -16.9), 'GT/CT': (-7.4, -21.2), 'AA/TC': (-1.6, -4.0), 'AC/TA': (-1.8, -3.8), 'CA/GC': (-2.6, -5.9), 'CC/GA': (-2.7, -6.0), 'GA/CC': (-5.0, -13.8), 'GC/CA': (-3.2, -7.1), 'TA/AC': (-2.3, -5.9), 'TC/AA': (-2.7, -7.0), 'AC/TT': (-0.9, -1.7), 'AT/TC': (-2.3, -6.3), 'CC/GT': (-3.2, -8.0), 'CT/GC': (-3.9, -10.6), 'GC/CT': (-4.9, -13.5), 'GT/CC': (-3.0, -7.8), 'TC/AT': (-2.5, -6.3), 'TT/AC': (-0.7, -1.2), 'AA/TG': (-1.9, -4.4), 'AG/TA': (-2.5, -5.9), 'CA/GG': (-3.9, -9.6), 'CG/GA': (-6.0, -15.5), 'GA/CG': (-4.3, -11.1), 'GG/CA': (-4.6, -11.4), 'TA/AG': (-2.0, -4.7), 'TG/AA': (-2.4, -5.8), 'AG/TT': (-3.2, -8.7), 'AT/TG': (-3.5, -9.4), 'CG/GT': (-3.8, -9.0), 'CT/GG': (-6.6, -18.7), 'GG/CT': (-5.7, -15.9), 'GT/CG': (-5.9, -16.1), 'TG/AT': (-3.9, -10.5), 'TT/AG': (-3.6, -9.8)}, 'imm_table': {'AG/TT': (1.0, 0.9), 'AT/TG': (-2.5, -8.3), 'CG/GT': (-4.1, -11.7), 'CT/GG': (-2.8, -8.0), 'GG/CT': (3.3, 10.4), 'GG/TT': (5.8, 16.3), 'GT/CG': (-4.4, -12.3), 'GT/TG': (4.1, 9.5), 'TG/AT': (-0.1, -1.7), 'TG/GT': (-1.4, -6.2), 'TT/AG': (-1.3, -5.3), 'AA/TG': (-0.6, -2.3), 'AG/TA': (-0.7, -2.3), 'CA/GG': (-0.7, -2.3), 'CG/GA': (-4.0, -13.2), 'GA/CG': (-0.6, -1.0), 'GG/CA': (0.5, 3.2), 'TA/AG': (0.7, 0.7), 'TG/AA': (3.0, 7.4), 'AC/TT': (0.7, 0.2), 'AT/TC': (-1.2, -6.2), 'CC/GT': (-0.8, -4.5), 'CT/GC': (-1.5, -6.1), 'GC/CT': (2.3, 5.4), 'GT/CC': (5.2, 13.5), 'TC/AT': (1.2, 0.7), 'TT/AC': (1.0, 0.7), 'AA/TC': (2.3, 4.6), 'AC/TA': (5.3, 14.6), 'CA/GC': (1.9, 3.7), 'CC/GA': (0.6, -0.6), 'GA/CC': (5.2, 14.2), 'GC/CA': (-0.7, -3.8), 'TA/AC': (3.4, 8.0), 'TC/AA': (7.6, 20.2), 'AA/TA': (1.2, 1.7), 'CA/GA': (-0.9, -4.2), 'GA/CA': (-2.9, -9.8), 'TA/AA': (4.7, 12.9), 'AC/TC': (0.0, -4.4), 'CC/GC': (-1.5, -7.2), 'GC/CC': (3.6, 8.9), 'TC/AC': (6.1, 16.4), 'AG/TG': (-3.1, -9.5), 'CG/GG': (-4.9, -15.3), 'GG/CG': (-6.0, -15.8), 'TG/AG': (1.6, 3.6), 'AT/TT': (-2.7, -10.8), 'CT/GT': (-5.0, -15.8), 'GT/CT': (-2.2, -8.4), 'TT/AT': (0.2, -1.5), 'AI/TC': (-8.9, -25.5), 'TI/AC': (-5.9, -17.4), 'AC/TI': (-8.8, -25.4), 'TC/AI': (-4.9, -13.9), 'CI/GC': (-5.4, -13.7), 'GI/CC': (-6.8, -19.1), 'CC/GI': (-8.3, -23.8), 'GC/CI': (-5.0, -12.6), 'AI/TA': (-8.3, -25.0), 'TI/AA': (-3.4, -11.2), 'AA/TI': (-0.7, -2.6), 'TA/AI': (-1.3, -4.6), 'CI/GA': (2.6, 8.9), 'GI/CA': (-7.8, -21.1), 'CA/GI': (-7.0, -20.0), 'GA/CI': (-7.6, -20.2), 'AI/TT': (0.49, -0.7), 'TI/AT': (-6.5, -22.0), 'AT/TI': (-5.6, -18.7), 'TT/AI': (-0.8, -4.3), 'CI/GT': (-1.0, -2.4), 'GI/CT': (-3.5, -10.6), 'CT/GI': (0.1, -1.0), 'GT/CI': (-4.3, -12.1), 'AI/TG': (-4.9, -15.8), 'TI/AG': (-1.9, -8.5), 'AG/TI': (0.1, -1.8), 'TG/AI': (1.0, 1.0), 'CI/GG': (7.1, 21.3), 'GI/CG': (-1.1, -3.2), 'CG/GI': (5.8, 16.9), 'GG/CI': (-7.6, -22.0), 'AI/TI': (-3.3, -11.9), 'TI/AI': (0.1, -2.3), 'CI/GI': (1.3, 3.0), 'GI/CI': (-0.5, -1.3)}, 'de_table': {'AA/.T': (0.2, 2.3), 'AC/.G': (-6.3, -17.1), 'AG/.C': (-3.7, -10.0), 'AT/.A': (-2.9, -7.6), 'CA/.T': (0.6, 3.3), 'CC/.G': (-4.4, -12.6), 'CG/.C': (-4.0, -11.9), 'CT/.A': (-4.1, -13.0), 'GA/.T': (-1.1, -1.6), 'GC/.G': (-5.1, -14.0), 'GG/.C': (-3.9, -10.9), 'GT/.A': (-4.2, -15.0), 'TA/.T': (-6.9, -20.0), 'TC/.G': (-4.0, -10.9), 'TG/.C': (-4.9, -13.8), 'TT/.A': (-0.2, -0.5), '.A/AT': (-0.7, -0.8), '.C/AG': (-2.1, -3.9), '.G/AC': (-5.9, -16.5), '.T/AA': (-0.5, -1.1), '.A/CT': (4.4, 14.9), '.C/CG': (-0.2, -0.1), '.G/CC': (-2.6, -7.4), '.T/CA': (4.7, 14.2), '.A/GT': (-1.6, -3.6), '.C/GG': (-3.9, -11.2), '.G/GC': (-3.2, -10.4), '.T/GA': (-4.1, -13.1), '.A/TT': (2.9, 10.4), '.C/TG': (-4.4, -13.1), '.G/TC': (-5.2, -15.0), '.T/TA': (-3.8, -12.6)}, 'dnac1': 50, 'dnac2': 0, 'selfcomp': False, 'saltcorr': 7, 'Na': 39, 'K': 75, 'Tris': 20, 'Mg': 10, 'dNTPs': 0}
2024-07-09 00:36:23,089 [INFO] Parameter: Tm_chem_correction_param_probe = {'DMSO': 0, 'fmd': 20, 'DMSOfactor': 0.75, 'fmdfactor': 0.65, 'fmdmethod': 1, 'GC': None}
2024-07-09 00:36:23,090 [INFO] Parameter: Tm_salt_correction_param_probe = None
2024-07-09 00:36:23,091 [INFO] Parameter: probe_GC_weight = 1
2024-07-09 00:36:23,094 [INFO] Parameter: probe_GC_content_min = 40
2024-07-09 00:36:23,096 [INFO] Parameter: probe_GC_content_opt = 50
2024-07-09 00:36:23,097 [INFO] Parameter: probe_GC_content_max = 60
2024-07-09 00:36:23,098 [INFO] Parameter: probeset_size_opt = 5
2024-07-09 00:36:23,099 [INFO] Parameter: probeset_size_min = 3
2024-07-09 00:36:23,100 [INFO] Parameter: max_graph_size = 5000
2024-07-09 00:36:23,101 [INFO] Parameter: n_sets = 100
2024-07-09 00:36:23,103 [INFO] Parameter: distance_between_probes = 0
2024-07-09 03:04:22,408 [INFO] Step - Set Selection: database contains 16129 oligos from 735 regions.
2024-07-09 03:04:22,449 [DEBUG] handle_msg[b011dd7910384d038ec20102cf492c08]({'header': {'date': datetime.datetime(2024, 7, 9, 1, 4, 22, 445000, tzinfo=tzutc()), 'msg_id': '75f170fe-5fbb-46a3-9620-3f145fbff019', 'msg_type': 'comm_msg', 'session': 'f15cb79e-4700-4ef8-aed8-65224aba1be6', 'username': '5bb12fba-c395-4a0a-afbc-31e5784dd153', 'version': '5.2'}, 'msg_id': '75f170fe-5fbb-46a3-9620-3f145fbff019', 'msg_type': 'comm_msg', 'parent_header': {}, 'metadata': {}, 'content': {'comm_id': 'b011dd7910384d038ec20102cf492c08', 'data': {'method': 'update', 'state': {'outputs': [{'output_type': 'display_data', 'data': {'text/plain': ' \x1b[35m100%\x1b[0m \x1b[36mFind Oligosets\x1b[0m \x1b[38;2;114;156;31m━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━\x1b[0m \x1b[32m806/806\x1b[0m \x1b[33m2:27:50\x1b[0m < \x1b[36m0:00:00\x1b[0m\n', 'text/html': '<pre style="white-space:pre;overflow-x:auto;line-height:normal;font-family:Menlo,\'DejaVu Sans Mono\',consolas,\'Courier New\',monospace"> <span style="color: #800080; text-decoration-color: #800080">100%</span> <span style="color: #008080; text-decoration-color: #008080">Find Oligosets</span> <span style="color: #729c1f; text-decoration-color: #729c1f">━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━</span> <span style="color: #008000; text-decoration-color: #008000">806/806</span> <span style="color: #808000; text-decoration-color: #808000">2:27:50</span> < <span style="color: #008080; text-decoration-color: #008080">0:00:00</span>\n</pre>\n'}, 'metadata': {}}]}, 'buffer_paths': []}}, 'buffers': []})
Select Gene Panel
In the probe database, the gene names are the keys of the database. All genes that do not have sufficient probes were removed from the database. Once we hve all genes with sufficient probes, we can run the gene set selection step. Therefore, we include additional metadata information to our adata object, i.e. the genes that have sufficient probes and the genes that fulfill both constraint (highly variable and sufficient probes).
[24]:
####### Load existing database #######
# dir_database = os.path.join(dir_output, "db_probes/4_db_probes_probesets")
# probe_database = OligoDatabase(min_oligos_per_region=min_probes_per_gene, write_regions_with_insufficient_oligos=True, lru_db_max_in_memory=n_jobs*2+2, database_name="db_probes", dir_output=dir_output, n_jobs=n_jobs)
# probe_database.load_database(dir_database=dir_database, region_ids=gene_ids, database_overwrite=True)
# get gene names of genes with sufficient number of probes to proceed with next step
genes_with_sufficient_probes = probe_database.database.keys()
# add extra clolumn to anndata to mark genes with sufficient probes
pbmc_data.var["sufficient_probes"] = False
for gene in highly_variable_genes:
if gene in genes_with_sufficient_probes:
pbmc_data.var["sufficient_probes"][gene] = True
# create a new variable that indicates if the gene passes the first constraint filter
pbmc_data.var["pass_constraints"] = [su_p and hi_v for su_p, hi_v in zip(pbmc_data.var["sufficient_probes"], pbmc_data.var["highly_variable"])]
ProbesetSelector class. We specify the number of genes n (20) and the keys in adata.obs and adata.var where we find the cell type annotations (celltype_key="celltype") and selected genes (celltype_key="celltype"), respectively.Executing the cell below will give us a warning that the cell type clusters for dendritic cells and megakaroycytes are quite small and therefore the genes that are selected to identify these cell types potentially don’t generalize very well. The method will not exclude these cell types automatically, but it can be done manually by setting the parameter celltypes to a subset of cell types instead of celltypes="all".
[25]:
## Select genes for gene panel
selector = sp.se.ProbesetSelector(pbmc_data, n=20, genes_key="pass_constraints", celltype_key="celltype", verbosity=1, save_dir=None)
selector.select_probeset()
selected_genes = selector.probeset.index[selector.probeset.selection]
Note: The following celltypes' test set sizes for forest training are below min_test_n (=20):
Dendritic cells : 9
Megakaryocytes : 3
The genes selected for those cell types potentially don't generalize well. Find the genes for each of those cell types in self.genes_of_primary_trees after running self.select_probeset().
/Users/lisa.barros/anaconda3/envs/odt_test/lib/python3.10/site-packages/threadpoolctl.py:1214: RuntimeWarning:
Found Intel OpenMP ('libiomp') and LLVM OpenMP ('libomp') loaded at
the same time. Both libraries are known to be incompatible and this
can cause random crashes or deadlocks on Linux when loaded in the
same Python program.
Using threadpoolctl may cause crashes or deadlocks. For more
information and possible workarounds, please see
https://github.com/joblib/threadpoolctl/blob/master/multiple_openmp.md
warnings.warn(msg, RuntimeWarning)
/Users/lisa.barros/anaconda3/envs/odt_test/lib/python3.10/site-packages/threadpoolctl.py:1214: RuntimeWarning:
Found Intel OpenMP ('libiomp') and LLVM OpenMP ('libomp') loaded at
the same time. Both libraries are known to be incompatible and this
can cause random crashes or deadlocks on Linux when loaded in the
same Python program.
Using threadpoolctl may cause crashes or deadlocks. For more
information and possible workarounds, please see
https://github.com/joblib/threadpoolctl/blob/master/multiple_openmp.md
warnings.warn(msg, RuntimeWarning)
/Users/lisa.barros/anaconda3/envs/odt_test/lib/python3.10/site-packages/threadpoolctl.py:1214: RuntimeWarning:
Found Intel OpenMP ('libiomp') and LLVM OpenMP ('libomp') loaded at
the same time. Both libraries are known to be incompatible and this
can cause random crashes or deadlocks on Linux when loaded in the
same Python program.
Using threadpoolctl may cause crashes or deadlocks. For more
information and possible workarounds, please see
https://github.com/joblib/threadpoolctl/blob/master/multiple_openmp.md
warnings.warn(msg, RuntimeWarning)
/Users/lisa.barros/anaconda3/envs/odt_test/lib/python3.10/site-packages/threadpoolctl.py:1214: RuntimeWarning:
Found Intel OpenMP ('libiomp') and LLVM OpenMP ('libomp') loaded at
the same time. Both libraries are known to be incompatible and this
can cause random crashes or deadlocks on Linux when loaded in the
same Python program.
Using threadpoolctl may cause crashes or deadlocks. For more
information and possible workarounds, please see
https://github.com/joblib/threadpoolctl/blob/master/multiple_openmp.md
warnings.warn(msg, RuntimeWarning)
/Users/lisa.barros/anaconda3/envs/odt_test/lib/python3.10/site-packages/threadpoolctl.py:1214: RuntimeWarning:
Found Intel OpenMP ('libiomp') and LLVM OpenMP ('libomp') loaded at
the same time. Both libraries are known to be incompatible and this
can cause random crashes or deadlocks on Linux when loaded in the
same Python program.
Using threadpoolctl may cause crashes or deadlocks. For more
information and possible workarounds, please see
https://github.com/joblib/threadpoolctl/blob/master/multiple_openmp.md
warnings.warn(msg, RuntimeWarning)
/Users/lisa.barros/anaconda3/envs/odt_test/lib/python3.10/site-packages/threadpoolctl.py:1214: RuntimeWarning:
Found Intel OpenMP ('libiomp') and LLVM OpenMP ('libomp') loaded at
the same time. Both libraries are known to be incompatible and this
can cause random crashes or deadlocks on Linux when loaded in the
same Python program.
Using threadpoolctl may cause crashes or deadlocks. For more
information and possible workarounds, please see
https://github.com/joblib/threadpoolctl/blob/master/multiple_openmp.md
warnings.warn(msg, RuntimeWarning)
/Users/lisa.barros/anaconda3/envs/odt_test/lib/python3.10/site-packages/threadpoolctl.py:1214: RuntimeWarning:
Found Intel OpenMP ('libiomp') and LLVM OpenMP ('libomp') loaded at
the same time. Both libraries are known to be incompatible and this
can cause random crashes or deadlocks on Linux when loaded in the
same Python program.
Using threadpoolctl may cause crashes or deadlocks. For more
information and possible workarounds, please see
https://github.com/joblib/threadpoolctl/blob/master/multiple_openmp.md
warnings.warn(msg, RuntimeWarning)
/Users/lisa.barros/anaconda3/envs/odt_test/lib/python3.10/site-packages/threadpoolctl.py:1214: RuntimeWarning:
Found Intel OpenMP ('libiomp') and LLVM OpenMP ('libomp') loaded at
the same time. Both libraries are known to be incompatible and this
can cause random crashes or deadlocks on Linux when loaded in the
same Python program.
Using threadpoolctl may cause crashes or deadlocks. For more
information and possible workarounds, please see
https://github.com/joblib/threadpoolctl/blob/master/multiple_openmp.md
warnings.warn(msg, RuntimeWarning)
[26]:
## Remove all genes from the database that are not selected for the gene panel
probe_database.database = {key: value for key, value in probe_database.database.items() if key in selected_genes}
probe_database.oligosets = {key: value for key, value in probe_database.oligosets.items() if key in selected_genes}
Get Final Padlock Probe Sequences
Once we have all selected genes, we create the final “ready to order” probe sequences. Calling the fuction below will produce two files, *[padlock, merfish, seqfish]_probes.yaml* and *[padlock, merfish, seqfish]_probes_order.yaml*. The latter file contains the ready to order probe sequences for each gene.
Parameters for Padlock Sequence Design
U_distance: preferred minimal distance between U(racils)detect_oligo_Tm_opt: optimal melting temperature of detection probetop_n_sets: maximum number of sets to report in *[padlock, merfish, seqfish]_probes.yaml* and *[padlock, merfish, seqfish]_probes_order.yaml*
Note: The melting temperature is used in 2 different stages (probe and detection oligo design), where a few parameters are shared and the others differ. Parameters for melting temperature -> for more information on parameters, see:here
[30]:
## Define parameters
U_distance = 5
detect_oligo_Tm_opt = 56
top_n_sets = 3
Tm_parameters_detection_oligo = {
"check": True, # default
"strict": True, # default
"c_seq": None, # default
"shift": 0, # default
"nn_table": getattr(mt, "DNA_NN3"), # Allawi & SantaLucia (1997)
"tmm_table": getattr(mt, "DNA_TMM1"), #default
"imm_table": getattr(mt, "DNA_IMM1"), #default
"de_table": getattr(mt, "DNA_DE1"), #default
"dnac1": 50, # [nM]
"dnac2": 0, # [nM]
"selfcomp": False, # default
"saltcorr": 7, # Owczarzy et al. (2008)
"Na": 39, # [mM]
"K": 0, # [mM] default
"Tris": 0, # [mM] default
"Mg": 0, # [mM] default
"dNTPs": 0, # [mM] default
}
Tm_chem_correction_param_detection_oligo = {
"DMSO": 0, # default
"fmd": 30,
"DMSOfactor": 0.75, # default
"fmdfactor": 0.65, # default
"fmdmethod": 1, # default
"GC": None, # default
}
Tm_salt_correction_param_detection_oligo = None
[32]:
## Design final sequences
probe_database = pipeline.design_final_probe_sequence(
oligo_database=probe_database,
min_thymines=min_thymines,
U_distance=U_distance,
detect_oligo_length_min=detect_oligo_length_min,
detect_oligo_length_max=detect_oligo_length_max,
detect_oligo_Tm_opt=detect_oligo_Tm_opt,
Tm_parameters_detection_oligo=Tm_parameters_detection_oligo,
Tm_chem_correction_param_detection_oligo=Tm_chem_correction_param_detection_oligo,
Tm_salt_correction_param_detection_oligo=Tm_salt_correction_param_detection_oligo,
Tm_parameters_probe=Tm_parameters_probe,
Tm_chem_correction_param_probe=Tm_chem_correction_param_probe,
Tm_salt_correction_param_probe=Tm_salt_correction_param_probe,
)
## Compute all required probe attributes for output
probe_database = pipeline.compute_probe_attributes(
oligo_database=probe_database,
Tm_parameters_probe=Tm_parameters_probe,
Tm_chem_correction_param_probe=Tm_chem_correction_param_probe,
Tm_salt_correction_param_probe=Tm_salt_correction_param_probe,
)
## Write output to files
pipeline.generate_output(oligo_database=probe_database, top_n_sets=top_n_sets)
2024-07-09 10:12:10,657 [DEBUG] handle_msg[1a931c4c89ec48e6be4917b9a4ac2f8f]({'header': {'date': datetime.datetime(2024, 7, 9, 8, 12, 10, 653000, tzinfo=tzutc()), 'msg_id': '394388fb-c687-478d-b79b-49e5fecd638c', 'msg_type': 'comm_msg', 'session': 'f15cb79e-4700-4ef8-aed8-65224aba1be6', 'username': '5bb12fba-c395-4a0a-afbc-31e5784dd153', 'version': '5.2'}, 'msg_id': '394388fb-c687-478d-b79b-49e5fecd638c', 'msg_type': 'comm_msg', 'parent_header': {}, 'metadata': {}, 'content': {'comm_id': '1a931c4c89ec48e6be4917b9a4ac2f8f', 'data': {'method': 'update', 'state': {'outputs': [{'output_type': 'display_data', 'data': {'text/plain': ' \x1b[35m100%\x1b[0m \x1b[36mDesign Final Padlock Sequence\x1b[0m \x1b[38;2;114;156;31m━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━\x1b[0m \x1b[32m20/20\x1b[0m \x1b[33m0:00:22\x1b[0m < \x1b[36m0:00:00\x1b[0m\n', 'text/html': '<pre style="white-space:pre;overflow-x:auto;line-height:normal;font-family:Menlo,\'DejaVu Sans Mono\',consolas,\'Courier New\',monospace"> <span style="color: #800080; text-decoration-color: #800080">100%</span> <span style="color: #008080; text-decoration-color: #008080">Design Final Padlock Sequence</span> <span style="color: #729c1f; text-decoration-color: #729c1f">━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━</span> <span style="color: #008000; text-decoration-color: #008000">20/20</span> <span style="color: #808000; text-decoration-color: #808000">0:00:22</span> < <span style="color: #008080; text-decoration-color: #008080">0:00:00</span>\n</pre>\n'}, 'metadata': {}}]}, 'buffer_paths': []}}, 'buffers': []})