[1]:
from IPython.display import HTML, display, Image
import yaml
import warnings
warnings.simplefilter('ignore')
import subprocess
import scanpy as sc
sc.settings.verbosity = 0
sc.logging.print_header()
import pandas as pd
#pd.set_option("max_columns", None) # show all cols
pd.set_option('max_colwidth', None) # show full width of showing cols
pd.set_option("expand_frame_repr", False) # print cols side by side as it's supposed to be
pd.options.display.max_seq_items = 200000
pd.options.display.max_rows = 400000
scanpy==1.10.1 anndata==0.10.7 umap==0.5.6 numpy==1.26.4 scipy==1.13.1 pandas==1.5.3 scikit-learn==1.5.0 statsmodels==0.14.2 igraph==0.11.5 pynndescent==0.5.12
End to end selection (short version)
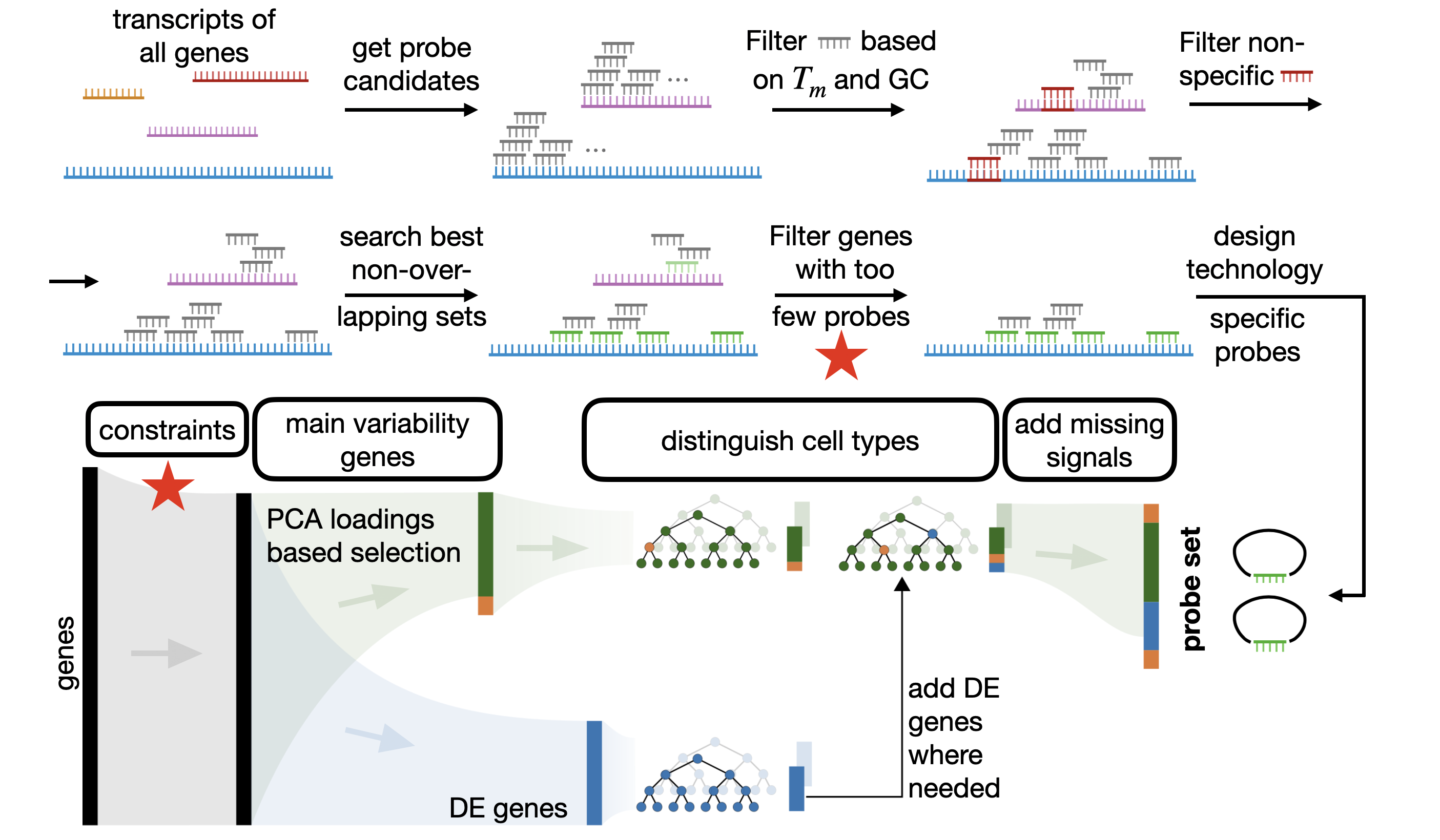
In this tutorial we showcase how to design probesets and select a suitable gene set with the spapros package. For all genes we design probes that fulfill experiment specific requirements and select only genes for which we can design sufficient probes. Spapros then selects genes that can distinguish the cell types in the data set and capture transcriptomic varation beyond cell type labels. The final probe sequences are designed in a last step for all selected genes. The figure below gives and
overview on the pipeline.
In this tutorial the full probe design is performed with one cli command. To understand the different steps in the pipeline we recommend to have a look at the long version of this tutorial.
[2]:
Image("../_tutorials/abstract_figures/end_to_end.png", width=900, embed=True)
[2]:
Import Packages
Besides spapros also install oligo_designer_toolsuite. Therefore, first setup a conda environment (packages is tested for Python 3.9 - 3.10), e.g.:
conda create -n odt python=3.10
conda activate odt
Then, install the required dependencies, i.e. Blast (2.15 or higher), BedTools (2.30 or higher), Bowtie (1.3 or higher) and Bowtie2 (2.5 or higher), that need to be installed independently. To install those tools via conda, please activate the Bioconda and conda-forge channels in your conda environment with and update conda and all packages in your environment:
conda config --add channels conda-forge
conda config --add channels bioconda
conda update --all
conda install "blast>=2.15.0"
conda install "bedtools>=2.30"
conda install "bowtie>=1.3.1"
conda install "bowtie2>=2.5"
All other required packages are automatically installed during the pip installation:
git clone https://github.com/HelmholtzAI-Consultants-Munich/oligo-designer-toolsuite.git
cd oligo-designer-toolsuite
git switch pipelines
pip install -e .
[3]:
import spapros as sp
print(f"spapros=={sp.__version__}")
spapros==0.1.5
Load and Preprocess Data
For this tutorial, we use a PBMC example scRNA-seq reference dataset. The count data should be log-normalised and genes should not be scaled to mean=0 and std=1. We can load the processed version of the data, including cell / gene filters, cell type annotations, and the umap embedding, directly with sp.ut.get_processed_pbmc_data() function. For a step by step processing of the PBMC dataset please refer to the basic usage tutorial. For sake
of simplicity, we pre-select the top 1000 highly variable genes for the probe and geneset selection. In real world applications we typically go for top 8000 genes.
[4]:
adata = sp.ut.get_processed_pbmc_data(n_hvg=1000)
highly_variable_genes = sorted(adata.var.loc[adata.var['highly_variable']].index.tolist())
print(f"Number of highly variable genes: {len(highly_variable_genes)}")
Number of highly variable genes: 1000
[5]:
# Save genes as a list in ./my_genes.txt -> if the file is renamed it needs to be changd in the config file as well
pd.DataFrame(highly_variable_genes).to_csv('./my_genes.txt', index=False, header=False)
Probe Design
Before we start with the probe design we have to create genomic region fasta files from which the probe database is created and which are used as background database for the specificity filter. Therefore, run the genomic_region_generator cli command of the oligo-designer-toolsuite from the terminal:
genomic_region_generator -c genomic_region_generator_ncbi.yaml
A default configuration file for the genomic region generator with NCBI annotations can be downloaded from here.
Note: if an error occurs for the unzipping of files, this might be due to a faulty download of files from the ftp server. In this case, try to download the files manually from the ftp server and use those files as input for the pipeline with custom input files. See ``spapros_tutorial_end_to_end_selection.ipynb`` tutorial for more information.
To conduct the probe design, run the *<technology>_probe_designer* cli command of the oligo-designer-toolsuite from the terminal:
scrinshot_probe_designer -c scrinshot_probe_designer.yaml
Alternativ pipelines: merfish_probe_designer, seqfish_probe_designer (see our resource table for an overview of differences between the technologies)
[6]:
cmd = "genomic_region_generator -c data/genomic_region_generator_ncbi.yaml"
process = subprocess.Popen(cmd, shell=True).wait()
2024-07-16 17:15:53,511 [INFO] Parameters Load Annotations:
2024-07-16 17:15:53,511 [INFO] source = ncbi
2024-07-16 17:15:53,511 [INFO] source_params = {'taxon': 'vertebrate_mammalian', 'species': 'Homo_sapiens', 'annotation_release': 110}
2024-07-16 17:23:33,702 [WARNING] /Users/lisa.barros/Desktop/oligo-designer-toolsuite/oligo_designer_toolsuite/utils/_sequence_parser.py:104: DtypeWarning: Columns (0) have mixed types. Specify dtype option on import or set low_memory=False.
csv_df = pd.read_csv(csv_file, sep="\t", names=self.GFF_HEADER, header=None)
2024-07-16 17:24:16,761 [INFO] The following annotation files are used for GTF annotation of regions: /Users/lisa.barros/Desktop/spapros/docs/_tutorials/output_genomic_region_generator_ncbi/annotation/GCF_000001405.40_GRCh38.p14_genomic.gtf and for fasta sequence file: /Users/lisa.barros/Desktop/spapros/docs/_tutorials/output_genomic_region_generator_ncbi/annotation/GCF_000001405.40_GRCh38.p14_genomic.fna .
2024-07-16 17:24:16,763 [INFO] The annotations are from NCBI source, for the species: Homo_sapiens, release number: 110 and genome assembly: GRCh38.p14
2024-07-16 17:28:18,217 [INFO] The genomic region 'exon' was stored in :/Users/lisa.barros/Desktop/spapros/docs/_tutorials/output_genomic_region_generator_ncbi/annotation/exon_annotation_source-NCBI_species-Homo_sapiens_annotation_release-110_genome_assemly-GRCh38.p14.fna.
2024-07-16 17:40:49,655 [INFO] The genomic region 'exon_exon_junction' was stored in :/Users/lisa.barros/Desktop/spapros/docs/_tutorials/output_genomic_region_generator_ncbi/annotation/exon_exon_junction_annotation_source-NCBI_species-Homo_sapiens_annotation_release-110_genome_assemly-GRCh38.p14.fna.
[7]:
cmd = "scrinshot_probe_designer -c data/scrinshot_probe_designer.yaml"
process = subprocess.Popen(cmd, shell=True).wait()
100% Database Loading ━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 887/887 1:09:20 < 0:00:00< 0:00:0504:28
100% Property Filter ━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 887/887 1:00:18 < 0:00:00< 0:00:0404:50
100% Specificity Filter: Exact Match ━━━━━━━━━━━━━━━ 887/887 0:04:31 < 0:00:00< 0:00:0100:26
100% Specificity Filter: Blastn Specificity ━━━━━━━━ 887/887 1:09:06 < 0:00:00< 0:00:1115:04
100% Specificity Filter: Blastn Crosshybridization ━━━━━ 810/… 0:02:… < 0:00:… < 0:00:…:00:…
100% Find Oligosets ━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 810/810 2:44:14 < 0:00:00< 0:00:0401:51
100% Design Final Padlock Sequence ━━━━━━━━━━━━━━━━━ 741/741 0:11:25 < 0:00:00< 0:00:0100:49
Load probe design filter and run gene panel selection
[8]:
##### Load probe design filter #####
genes_without_enough_probes = pd.read_csv('output_scrinshot_probe_designer/db_probes/regions_with_insufficient_oligos_for_db_probes.txt', index_col=0, sep="\t").index.tolist()
adata.var["has_enough_probes"] = [g not in genes_without_enough_probes for g in adata.var_names]
adata.var["pass_constraints"] = adata.var["has_enough_probes"] & adata.var["highly_variable"]
[9]:
##### Select genes for gene panel #####
selector = sp.se.ProbesetSelector(adata, n=20, genes_key="pass_constraints", celltype_key="celltype", verbosity=1, save_dir=None)
selector.select_probeset()
selected_genes = selector.probeset.index[selector.probeset.selection]
Note: The following celltypes' test set sizes for forest training are below min_test_n (=20):
Dendritic cells : 9
Megakaryocytes : 3
The genes selected for those cell types potentially don't generalize well. Find the genes for each of those cell types in self.genes_of_primary_trees after running self.select_probeset().
/Users/lisa.barros/anaconda3/envs/odt_test/lib/python3.10/site-packages/threadpoolctl.py:1214: RuntimeWarning:
Found Intel OpenMP ('libiomp') and LLVM OpenMP ('libomp') loaded at
the same time. Both libraries are known to be incompatible and this
can cause random crashes or deadlocks on Linux when loaded in the
same Python program.
Using threadpoolctl may cause crashes or deadlocks. For more
information and possible workarounds, please see
https://github.com/joblib/threadpoolctl/blob/master/multiple_openmp.md
warnings.warn(msg, RuntimeWarning)
/Users/lisa.barros/anaconda3/envs/odt_test/lib/python3.10/site-packages/threadpoolctl.py:1214: RuntimeWarning:
Found Intel OpenMP ('libiomp') and LLVM OpenMP ('libomp') loaded at
the same time. Both libraries are known to be incompatible and this
can cause random crashes or deadlocks on Linux when loaded in the
same Python program.
Using threadpoolctl may cause crashes or deadlocks. For more
information and possible workarounds, please see
https://github.com/joblib/threadpoolctl/blob/master/multiple_openmp.md
warnings.warn(msg, RuntimeWarning)
/Users/lisa.barros/anaconda3/envs/odt_test/lib/python3.10/site-packages/threadpoolctl.py:1214: RuntimeWarning:
Found Intel OpenMP ('libiomp') and LLVM OpenMP ('libomp') loaded at
the same time. Both libraries are known to be incompatible and this
can cause random crashes or deadlocks on Linux when loaded in the
same Python program.
Using threadpoolctl may cause crashes or deadlocks. For more
information and possible workarounds, please see
https://github.com/joblib/threadpoolctl/blob/master/multiple_openmp.md
warnings.warn(msg, RuntimeWarning)
/Users/lisa.barros/anaconda3/envs/odt_test/lib/python3.10/site-packages/threadpoolctl.py:1214: RuntimeWarning:
Found Intel OpenMP ('libiomp') and LLVM OpenMP ('libomp') loaded at
the same time. Both libraries are known to be incompatible and this
can cause random crashes or deadlocks on Linux when loaded in the
same Python program.
Using threadpoolctl may cause crashes or deadlocks. For more
information and possible workarounds, please see
https://github.com/joblib/threadpoolctl/blob/master/multiple_openmp.md
warnings.warn(msg, RuntimeWarning)
/Users/lisa.barros/anaconda3/envs/odt_test/lib/python3.10/site-packages/threadpoolctl.py:1214: RuntimeWarning:
Found Intel OpenMP ('libiomp') and LLVM OpenMP ('libomp') loaded at
the same time. Both libraries are known to be incompatible and this
can cause random crashes or deadlocks on Linux when loaded in the
same Python program.
Using threadpoolctl may cause crashes or deadlocks. For more
information and possible workarounds, please see
https://github.com/joblib/threadpoolctl/blob/master/multiple_openmp.md
warnings.warn(msg, RuntimeWarning)
/Users/lisa.barros/anaconda3/envs/odt_test/lib/python3.10/site-packages/threadpoolctl.py:1214: RuntimeWarning:
Found Intel OpenMP ('libiomp') and LLVM OpenMP ('libomp') loaded at
the same time. Both libraries are known to be incompatible and this
can cause random crashes or deadlocks on Linux when loaded in the
same Python program.
Using threadpoolctl may cause crashes or deadlocks. For more
information and possible workarounds, please see
https://github.com/joblib/threadpoolctl/blob/master/multiple_openmp.md
warnings.warn(msg, RuntimeWarning)
/Users/lisa.barros/anaconda3/envs/odt_test/lib/python3.10/site-packages/threadpoolctl.py:1214: RuntimeWarning:
Found Intel OpenMP ('libiomp') and LLVM OpenMP ('libomp') loaded at
the same time. Both libraries are known to be incompatible and this
can cause random crashes or deadlocks on Linux when loaded in the
same Python program.
Using threadpoolctl may cause crashes or deadlocks. For more
information and possible workarounds, please see
https://github.com/joblib/threadpoolctl/blob/master/multiple_openmp.md
warnings.warn(msg, RuntimeWarning)
/Users/lisa.barros/anaconda3/envs/odt_test/lib/python3.10/site-packages/threadpoolctl.py:1214: RuntimeWarning:
Found Intel OpenMP ('libiomp') and LLVM OpenMP ('libomp') loaded at
the same time. Both libraries are known to be incompatible and this
can cause random crashes or deadlocks on Linux when loaded in the
same Python program.
Using threadpoolctl may cause crashes or deadlocks. For more
information and possible workarounds, please see
https://github.com/joblib/threadpoolctl/blob/master/multiple_openmp.md
warnings.warn(msg, RuntimeWarning)
[10]:
##### Get probes of genes #####
with open('output_scrinshot_probe_designer/padlock_probes_order.yml', 'r') as file:
all_probes = yaml.safe_load(file)
probes = {g: all_probes[g] for g in selected_genes}
[11]:
print(f"Selected genes: {sorted(list(probes.keys()))}")
probes["CTSS"]
Selected genes: ['CD79A', 'CST3', 'CTSS', 'FCER1G', 'FCGR3A', 'FCN1', 'FGFBP2', 'GNLY', 'GPX1', 'GRN', 'GZMA', 'GZMH', 'GZMK', 'IL32', 'LYAR', 'MAL', 'MS4A1', 'PF4', 'PRF1', 'TYROBP']
[11]:
{'oligoset_1': {'CTSS::21801': {'sequence_padlock_probe': 'CAGCAGTTGCTCCCACAGTTCCTCTATGATTACTGACTGCGTCTATTTAGTGGAGCCTTCTCCTATCTTCTTTACCAGCCGTTTCATTGTGATAGAAC',
'sequence_detection_oligo': 'CCGTTTCATTGTGATAGAACCAGCAGTTGCUCCCACAGU[fluorophore]'},
'CTSS::3064': {'sequence_padlock_probe': 'GCCACAGCTTCTTTCAGGACATCCTCTATGATTACTGACTGCGTCTATTTAGTGGAGCCTTCTCCTATCTTCTTTCCAACAGACACTGGGCCTTTATTG',
'sequence_detection_oligo': 'CAGACACTGGGCCTTTATTGGCCACAGCUTCTTUCAGGAC[fluorophore]'},
'CTSS::22579': {'sequence_padlock_probe': 'GCGTCTGAGTCGATGCCCTTTCCTCTATGATTACTGACTGCGTCTATTTAGTGGAGCCTTCTCCTATCTTCTTTATCCATGGCTTTGTAGGGATAGGAA',
'sequence_detection_oligo': 'TGGCTTTGTAGGGATAGGAAGCGTCTGAGTCGAUGCCCU[fluorophore]'},
'CTSS::979': {'sequence_padlock_probe': 'TTCCCATTGAATGCTCCAGGTTCCTCTATGATTACTGACTGCGTCTATTTAGTGGAGCCTTCTCCTATCTTCTTTTGCCCAGATCGTATGAGTGCA',
'sequence_detection_oligo': 'GCCCAGATCGTATGAGTGCATTCCCATUGAATGCUCCAGG[fluorophore]'},
'CTSS::22236': {'sequence_padlock_probe': 'AGCACCACAAGAACCCATGTCTTCCTCTATGATTACTGACTGCGTCTATTTAGTGGAGCCTTCTCCTATCTTCTTTACAGCACTGAAAGCCCAGCA',
'sequence_detection_oligo': 'ACAGCACUGAAAGCCCAGCAAGCACCACAAGAACCCATGU[fluorophore]'}},
'oligoset_2': {'CTSS::21801': {'sequence_padlock_probe': 'CAGCAGTTGCTCCCACAGTTCCTCTATGATTACTGACTGCGTCTATTTAGTGGAGCCTTCTCCTATCTTCTTTACCAGCCGTTTCATTGTGATAGAAC',
'sequence_detection_oligo': 'CCGTTTCATTGTGATAGAACCAGCAGTTGCUCCCACAGU[fluorophore]'},
'CTSS::22579': {'sequence_padlock_probe': 'GCGTCTGAGTCGATGCCCTTTCCTCTATGATTACTGACTGCGTCTATTTAGTGGAGCCTTCTCCTATCTTCTTTATCCATGGCTTTGTAGGGATAGGAA',
'sequence_detection_oligo': 'TGGCTTTGTAGGGATAGGAAGCGTCTGAGTCGAUGCCCU[fluorophore]'},
'CTSS::979': {'sequence_padlock_probe': 'TTCCCATTGAATGCTCCAGGTTCCTCTATGATTACTGACTGCGTCTATTTAGTGGAGCCTTCTCCTATCTTCTTTTGCCCAGATCGTATGAGTGCA',
'sequence_detection_oligo': 'GCCCAGATCGTATGAGTGCATTCCCATUGAATGCUCCAGG[fluorophore]'},
'CTSS::2942': {'sequence_padlock_probe': 'GGCCACAGCTTCTTTCAGGACTCCTCTATGATTACTGACTGCGTCTATTTAGTGGAGCCTTCTCCTATCTTCTTTCCAACAGACACTGGGCCTTTATT',
'sequence_detection_oligo': 'ACAGACACTGGGCCTTTATTGGCCACAGCUTCTTUCAGGA[fluorophore]'},
'CTSS::22236': {'sequence_padlock_probe': 'AGCACCACAAGAACCCATGTCTTCCTCTATGATTACTGACTGCGTCTATTTAGTGGAGCCTTCTCCTATCTTCTTTACAGCACTGAAAGCCCAGCA',
'sequence_detection_oligo': 'ACAGCACUGAAAGCCCAGCAAGCACCACAAGAACCCATGU[fluorophore]'}},
'oligoset_3': {'CTSS::21801': {'sequence_padlock_probe': 'CAGCAGTTGCTCCCACAGTTCCTCTATGATTACTGACTGCGTCTATTTAGTGGAGCCTTCTCCTATCTTCTTTACCAGCCGTTTCATTGTGATAGAAC',
'sequence_detection_oligo': 'CCGTTTCATTGTGATAGAACCAGCAGTTGCUCCCACAGU[fluorophore]'},
'CTSS::2173': {'sequence_padlock_probe': 'GTCTGAGTCGATGCCCTTGTTTCCTCTATGATTACTGACTGCGTCTATTTAGTGGAGCCTTCTCCTATCTTCTTTATGGCTTTGTAGGGATAGGAAGC',
'sequence_detection_oligo': 'GCTTTGTAGGGATAGGAAGCGTCTGAGTCGAUGCCCTTGU[fluorophore]'},
'CTSS::979': {'sequence_padlock_probe': 'TTCCCATTGAATGCTCCAGGTTCCTCTATGATTACTGACTGCGTCTATTTAGTGGAGCCTTCTCCTATCTTCTTTTGCCCAGATCGTATGAGTGCA',
'sequence_detection_oligo': 'GCCCAGATCGTATGAGTGCATTCCCATUGAATGCUCCAGG[fluorophore]'},
'CTSS::2942': {'sequence_padlock_probe': 'GGCCACAGCTTCTTTCAGGACTCCTCTATGATTACTGACTGCGTCTATTTAGTGGAGCCTTCTCCTATCTTCTTTCCAACAGACACTGGGCCTTTATT',
'sequence_detection_oligo': 'ACAGACACTGGGCCTTTATTGGCCACAGCUTCTTUCAGGA[fluorophore]'},
'CTSS::22236': {'sequence_padlock_probe': 'AGCACCACAAGAACCCATGTCTTCCTCTATGATTACTGACTGCGTCTATTTAGTGGAGCCTTCTCCTATCTTCTTTACAGCACTGAAAGCCCAGCA',
'sequence_detection_oligo': 'ACAGCACUGAAAGCCCAGCAAGCACCACAAGAACCCATGU[fluorophore]'}}}